

 当前位置:首页 > 专题 > 分子生物学
当前位置:首页 > 专题 > 分子生物学
分子克隆技术又称基因克隆或DNA重组技术,是在体外对DNA分子按照既定的目的和方案进行人工重组,然后将重组分子(目的基因或DNA片段与合适的载体连接)导入到合适的受体细胞中,使其在细胞中复制与扩增,以获得多拷贝的DNA分子,并使受体细胞获得新的遗传特征的过程。
1. 目的基因克隆
从特定的生物基因组或cDNA中分离和扩大繁殖获得足够量的目的基因或 cDNA序列。
2. 载体的准备
选择合适的载体,并对载体DNA进行克隆,制备足够量的达到一定纯度的载体。
3. 目的基因与载体的酶切、连接
选择适当的克隆策略,将目的基因连接于载体的多克隆位点或启动子和终止子之间,实现 DNA 重组。
4. 重组DNA转化/转染/转导
将重组DNA转化/转染/转导进入大肠杆菌细胞、酵母菌细胞、植物外植体或动物细胞。
5. 重组体的筛选与鉴定
利用载体上提供的选择标记基因进行筛选,获得具有重组DNA分子的阳性克隆,或通过植株再生、胚移植等手段获得转基因植株或转基因动物。还需对所获得的转化细胞、转基因植株或转基因动物进行分子鉴定。
6. 重组体的大量培养,外源基因表达效应分析与开发应用
将经筛选和鉴定出来的大肠杆菌、酵母转化细胞进行大量扩大繁殖,对外源基因的表达蛋白进行分离与纯化并进行后续结构与功能的研究。
载体是用于携带重组DNA,并且能够使外源DNA一起复制与表达的运载工具。
质粒是一种独立于宿主细胞染色体以外,能独立自主复制的遗传单位。它们常见的形式是细菌中环状的双链DNA分子,有的也存在于古细菌和真核生物中,其大小在1~200kb不等。质粒是“裸”DNA,不编码必要的基因,在自然界中,质粒通常携带有利于有机体生存的基因,并赋予诸如抗生素耐药性等选择优势。
质粒在遗传工程中占有重要的位置,它被认为是复制子,能够在合适的宿主内自主复制的DNA单位,被广泛用作分子克隆的载体,驱动宿主体内重组DNA序列的复制,在实验室中,质粒可以通过转化进入细胞。
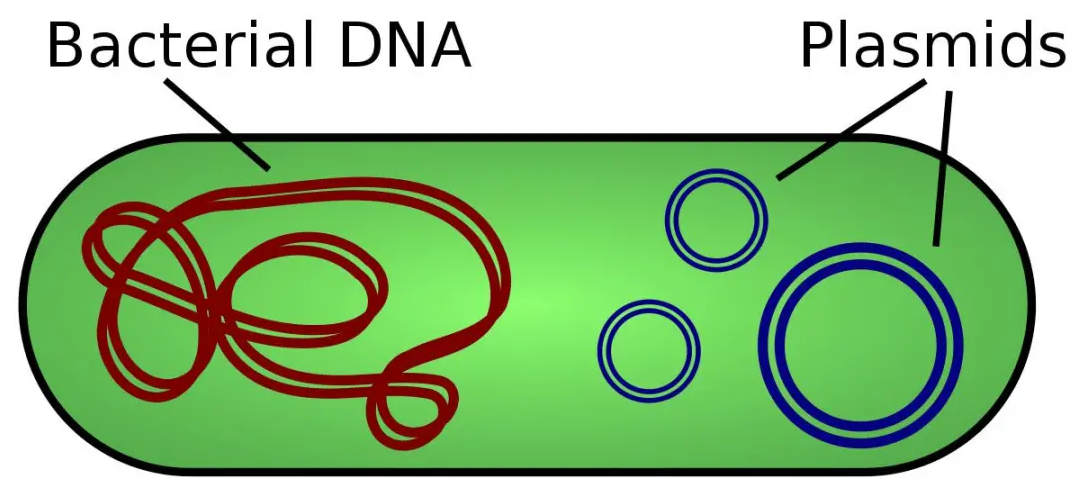
质粒可以按照多种方式进行分类。
1、按照质粒能否通过细菌的接合作用,可分为接合性质粒和非接合性质粒。接合质粒含有促进不同细胞间性结合的转移基因,在复杂的接合过程中,质粒可通过某些转移基因编码的性菌毛从一个细菌转移到另一个细菌,非接合质粒不能起始接合,因此只能借助接合质粒进行转移。
2、根据质粒的不相容性,可分为不相容性和相容性。一个微生物可以容纳不同类型的质粒,但不同的质粒只有在相容的情况下才能存在于一个细菌细胞中。如果两个质粒不相容,其中一个或另一个会迅速从细胞中消失。
3、根据复制性质,可分为严紧控制型和松弛控制型。严紧控制复制型质粒的复制酶系与染色体DNA复制共用,只能在细胞周期的一定阶段进行复制,当细胞染色体停止复制时,质粒也就不再复制。松弛控制复制型的质粒的复制酶系不受染色体DNA复制酶系的影响,在整个细胞生长周期中随时都可以复制,在染色体复制已经停止时质粒仍能继续复制。
4、此外,根据质粒的功能不同,质粒还有如下的分类,如致育质粒、抗性质粒、Col质粒、降解质粒、侵入性质粒。
用于携带DNA片段进入宿主细胞进行复制或保存的载体必须满足以下基本条件:①具有对受体细胞的可转移性,能携带外源其因进入宿主细胞;②能在宿主细胞中自主复制,并实现外源基因的增殖;③具有由单一限制性内切核酸酶识别位点组成的多克隆位点(mulfiple cloning site,MCS),可供外源基因的插入;④具有合适的选择性标记,用于含有重组质粒的宿主细胞筛选。
天然质粒往往存在这样或那样的缺陷,不适宜直接用作克隆载体,常需要进行人工改造。结合上述条件,这些改造也就主要集中在以下几个方面∶
(1)选择合适的复制起始位点。为了使构建的质粒克隆载体能在受体细胞中进行有效复制,且获得足够数量的拷贝数,需要改变复制起始位点,一般选择组装松散型质粒复制起始位点。
(2)加入合适的选择标记基因。为便于重组子筛选,需要在质粒克隆载体上加入合适的标记基因,主要有β-半乳糖苷酶基因(lacZ)和抗生素抗性基因。前者用于克隆子蓝、白斑筛选;常用的抗生素抗性基因主要有氨苄青霉素抗性其因(ampicillin resistance gene,ampr)、四环素抗性基因(tetracycline resistance gene,tetr)、氯霉素抗性基因(chloramphenicol resistance gene,cmr)、卡那霉素抗性其因(kanamycin resistance gene,kanr)和新霉素抗性基因(neomycin resistance gene,neor)等。
(3)增加或减少酶切位点。质粒载体利用限制性内切核酸酶识别位点来作为外源DNA片段插入的克隆位点,这种位点必须是单一酶切位点,而且数量越多越便于多种类型末端的DNA插入。可通过删除天然质粒中的部分重复的限制性内切核酸酶识别位点使之成为单一酶切位点,也可通过增加新的单一限制性内切核酸酶位点,在特定的部位(如在筛选标记基因上)组装一个含有多种单一限制性内切核酸酶识别序列的多克隆位点(MCS)。
(4)缩短长度。质粒转化受体细胞的效率同质粒DNA分子大小相关,小分子质量质粒转化效率较高。可通过切去质粒上不必要的片段,提高转化宿主细胞的效率,提高其外源 DNA 片段的装载量。
经过改进和创新的克隆载体越来越小、容量越来越大、工作效率越来越高、使用也越来越方便。
病毒载体是指以病毒为基础的基因载体,具体方法是对病毒基因组进行操作和改造,使它携带外源基因和相关基因元件,并被包装成病毒颗粒,病毒载体是一种常见的分子生物学工具。用于基因导入并能在临床开展研究的病毒载体一般应具备以下基本条件:
①携带外源基因并能包装成病毒颗粒;
②介导外源基因的转移和表达;
③对人体不致病;
④在环境中不会引起增殖和传播。
非病毒载体一般是指质粒DNA,也可以是无载体的核酸,如反义寡核苷酸、Ribozyme、siRNA等,它是利用非病毒的载体材料的物化性质来介导基因的转移。
(含原核和真核2个复制子,能够在原核和真核两种宿主细胞中复制,并可以在真核细胞中有效表达)。
克隆载体:具有克隆载体的基本元件(Ori,Ampr,MCS等),可以携带DNA片段或外源基因进入受体细胞并克隆和大量扩增DNA片段(基因)的载体。
表达载体:克隆载体中加入一些与表达调控(具有转录/翻译所必需的DNA顺序)有关的元件即称为表达载体。
原核表达载体即能携带插入的外源基因序列进入原核细胞中进行表达的载体。
①启动子
启动子的强弱是影响表达量的决定因素之一,原核表达载体启动子主要有lac、 trp、tac、T7噬菌体启动子和IPL启动子。乳糖操纵子中lac启动子的转录可通过CAP和cAMP来激活,又被调节基因产生的阻遏蛋白与操纵基因结合所阻止。乳糖类似物 IPTG可与阻遏蛋白结合解除阻碍,因此可用IPTG来调控 lac启动子的表达。色氨酸启动子trp的阻遏蛋白须与色氨酸结合才有活性。tac是由lac和trp构建的杂合启动子,受IPTG的诱导,启动能力比lac和trp强。T7噬菌体启动子具有高度特异性,只有T7 RNA聚合酶才能使其启动,而且T7 RNA聚合酶比大肠杆菌聚合酶活性高得多,可用来高效表达基因。IPL是噬菌体的早期左向启动子,活性比trp高11倍,受温度的调控。
②转录终止子
在构建表达载体时,为了稳定载体系统,防止克隆的外源基因表达干扰载体的稳定性,一般在多克隆位点的下游插入一段转录终止子。
③核糖体结合位点
转录出的mRNA必须借助宿主细胞的蛋白质合成系统翻译出目的蛋白。细胞中的核糖体必须在mRNA上找到有效核糖体结合位点以及其中的SD(Shine-Dalgarno)序列,从而启动临近的翻译起始密码子的蛋白质翻译。核糖体结合位点可由目的基因自己带入,也可利用载体预先装载(一般要与ATG相隔3~11 bp)。
①组成型表达 T7噬菌体启动子等属于组成型启动子,在该类启动子的驱动下,外源基因源源不断地表选。组成型表达通常产量高,但不适合表达一些对宿主细胞有害的蛋白。
②诱导型表达 lac及其衍生tac启动子都是IPTG诱导型启动子,trp是色氨酸诱导型启动子,IPL的表达受温度调控。这些诱导型启动子的可控性表达,既可避免表达产物对宿主前期生长的不利影响,又可减少基因表达产物遭受蛋白酶的降解作用,特别适合有毒蛋自的表达。
③融合型表达 表达载体的多克隆位点上有一段融合表达标签(tag),表达为融合蛋白(N端融合或C端融合),可方便后续的蛋白分离纯化或检测。常见的融合表达标签有谷胱甘肽S转移酶基因(GST)标签或6×His标签。对于特别小的目的蛋自,使用分子质量较大的GST标签为好。
④分泌型表达 在起始密码和目的基因之间加入一个信号肽,可以引导目的蛋白穿越细胞膜。可避免表达产物在细胞内过度积累而影响细胞的生长,或者形成包含体,而且表达产物是可溶性的,不需要复性。
由于原核生物和真核生物存在糖基化、酰基化等翻译后修饰反应机制的差异,真核基因的原核表达难以获得有活性的蛋白,酵母成为了真核表达的首选系统。
酵母表达载体多数从pBR322基本骨架衍生而来,含有该质粒复制起始位点(ori),lacZ基因、bla或ampr、tetr等基因。启动子有GAP,AOX1、AUG1和GAL1等.其中,GAP是组成型启动子,AOX1,AUG1和GAL1分别受甲醇、半乳糖和维生素B1等诱导表达的诱导型启动子。
酵母表达载体多为穿梭载体,既可以在大肠杆菌中繁殖,获得足够的载体DNA进行体外操作,又可以在酵母中复制、表达和选择。
Ti质粒(tumor inducing plasmid)是根据农杆菌中发现的可引发植物产生冠瘿瘤的质粒。Ti质粒可分为T-DNA、Vir、Con 和Ori 4个功能区。
利用T-DNA可携带外源DNA片段并整合到植物基因组的特性,经对天然Ti质粒的改造,用于构建植物遗传转化的表达载体,分为一元表达载体和双元表达载体等类型。
这类载体是由质粒作为基本骨架构建而成,带有原核复制区、选择性标记基因、启动子、polyA尾信号序列、动物细胞中的选择基因[neor、胸苷激酶基因(tk)、绿色荧光蛋白基因(gfp)和腺嘌呤磷酸核糖转移酶基因(aprt)]等有关元件。山羊乳腺特异性表达载体(pBC1)是典型的动物质粒表达载体。
病毒表达载体是以病毒基因组序列为基础,插入必要的表达载体元件所构建成的真核基因转移工具。这些表达载体元件有①源于病毒的启动子和增强子;②病毒的复制子序列以及所需的反式作用因子编码基因;③poly A加尾信号。
病毒表达载体可用于∶①外源蛋白的真核表达;②基因治疗;③多价、多联活载体疫苗的研制;④基因功能的鉴定
重组型病毒载体
以完整的病毒基因组为改造对象,一方面选择性地删除病毒的某些必需基因,缺失的必需基因的功能由互补细胞反式提供;另一方面用外源基因表达单位替代病毒非必需基因区;而病毒复制和包装所需的顺式作用元件不变。这类载体一般通过同源重组方法将外源基因表达单位插入病毒基因组中。
无病毒基因的病毒载体
这类载体系统往往由载体质粒和辅助系统组成。重组载体质粒主要由外源基因表达盒、病毒复制和包装所必需的顺式作用元件及质粒骨架组成。辅助系统包括病毒复制和包装所必需的所有反式作用元件。在辅助系统的作用下,重组载体质粒(包含或不包含质粒骨架)以特定形式(单链或双链,DNA或 RNA)被包装到病毒壳粒中,其中不含有任何病毒基因。
这类载体的优点在于载体病毒本身安全性好,容量大;缺点在于往往需要辅助病毒参与载体DNA的包装,而辅助病毒又难以同载体病毒分离开来,造成最终产品中辅助病毒污染,从而影响其应用。实际上,无病毒基因的病毒载体可以看作是重组病毒载体的一种极端减毒情况。重组AAV 载体就属于无病毒基因的病毒载体。
腺病毒是一种无包膜的双链DNA病毒,在自然界中广泛分布。完整的病毒颗粒为二十面体对称结构,直径70-100nm,衣壳含有240个六邻体(hexon)、12个五邻体(penton)、12根纤毛(fiber)及一些小蛋白等。哺乳动物腺病毒的基因组DNA长约36kb,基因组的两端各有约100bp的反向末端重复序列(ITR),ITR与末端蛋白(TP)相结合,与基因组复制及早期基因的转录有关,ITR的内侧为病毒包装信号Ψ,参与腺病毒基因组的衣壳化。
基于人血清5型腺病毒的基因组结构,结合各基因的功能,科学家们开发了缺失E1和E3基因的Ad5腺病毒载体。E1基因在组装感染性病毒颗粒时必不可少,但是可以在HEK293包装细胞中得到补充,而E3基因不影病毒的包装。由于E1和E3基因的缺失,腺病毒载体可插入高达7.5kb的外源基因。
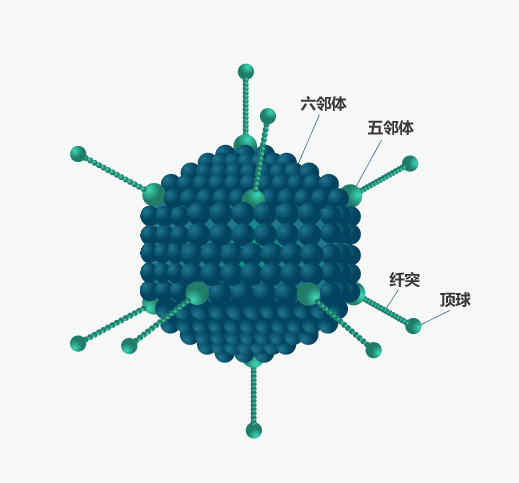
腺病毒感染细胞的原理
腺病毒通过其纤毛蛋白C端的球状结构域与细胞表面特异性受体(Ad5腺病毒特异性识别细胞表面的CAR受体;Ad5/F35腺病毒能特异性识别细胞表面的CD46受体)结合,同时病毒的五邻体蛋白与细胞表面的整联蛋白αvβ3和αvβ5相互作用,通过细胞内吞完成病毒的内化。
关于E1与E3基因
E1基因是一种早期最先启动的基因,对于腺病毒其他早期基因的转录是必需的,因此缺失E1的腺病毒载体不能有效复制和产生各种病毒蛋白,从而不能完成病毒生活周期。腺病毒的E3区编码5-9个蛋白,主要功能都是帮助受染细胞逃逸机体免疫系统的识别和清除。
Ad5与Ad5/F35对比
Ad5腺病毒的高效感染依赖于靶细胞膜上的CAR和αvβ整合素,但是一些免疫细胞的细胞膜上却缺乏这些受体,如造血干细胞。这就使得研究人员寻找新的腺病毒血清型,最终发现F35血清型对造血干细胞有很强的靶向性,因此,嵌合型Ad5/F35应用而生。下面是Ad5 & Ad5/F35的结构示意图和区别:
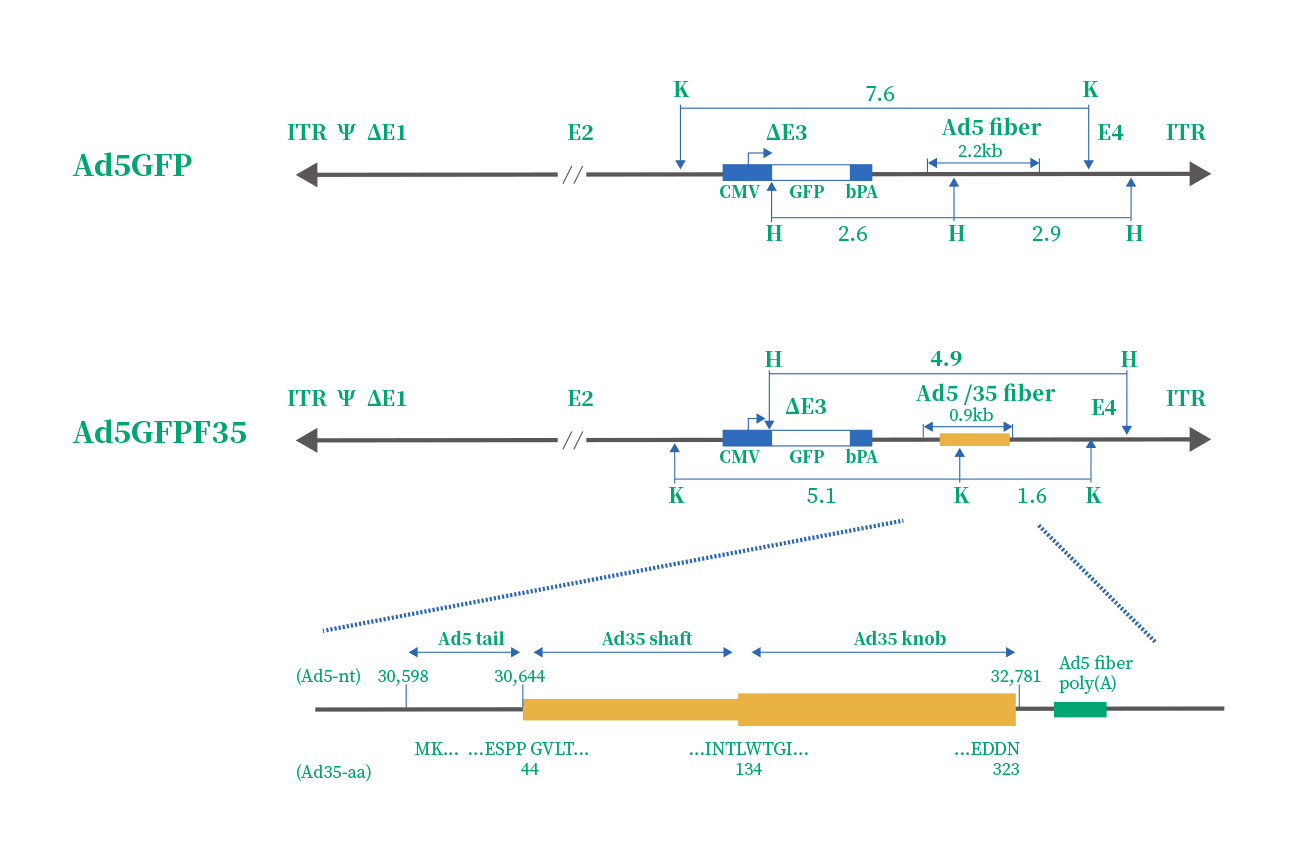
| 腺病毒种类 | 载体特点 | 功能 | 感染细胞受体 | 包装系统 |
|---|---|---|---|---|
| AdV5 | 缺失E1和E3基因,允许插入长约8kb的外源基因 |
除免疫、血液类细胞外, 感染大部分细胞 |
CAR | AdMax |
| AdV5/F35 |
一个嵌合型腺病毒载体,主要结构是在AdV5的基础上,其受体结合位置的纤突(Fiber)改造成了F35型腺病毒(F35)的纤突(纤毛蛋白的Knob和shaft)。其细胞受体从AdV5的CAR变成了CD46,其他的基因仍旧保留AdV5载体的特性,能有效转导CAR表达不足的多种重要靶细胞,尤其是对肿瘤细胞及造型干细胞、间充质干细胞等有较高的感染效率。 |
理论上感染所有有细胞核的细胞 |
CD46 | AdMax |
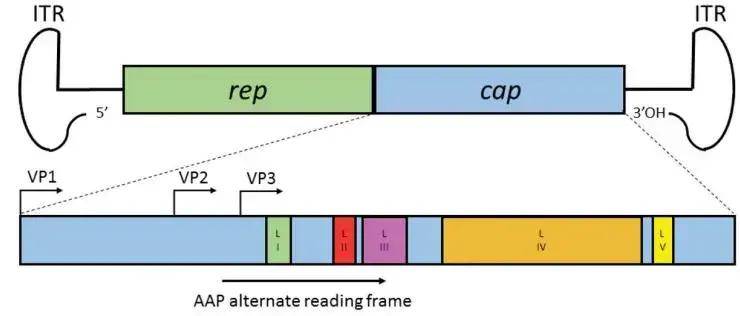
腺相关病毒(Adeno-associated virus,AAV)是一种单链DNA复制缺陷型细小病毒,它的生命周期依赖于复制病毒的参与。AAV基因组大小为4.7 kb,由末端反向重复序列ITR和中间的rep、cap基因组成。ITRs对于病毒的复制和包装具有重要作用;rep基因编码参与病毒复制、包装和基因组整合的非结构蛋白,cap基因编码结构蛋白VP1、VP2和VP3,3种蛋白分别按1:1:10的比例组装形成病毒衣壳,充当病毒基因传递载体。此外,cap基因内嵌套的另一个开放阅读框编码装配激活蛋白AAP,参与衣壳蛋白的靶向和装配。
AAV转导细胞主要经历了细胞表面受体介导的内吞、逃离内体、入核、脱衣壳和双链转化几个过程。进入细胞后,可以在辅助病毒的存在下进入其复制周期。
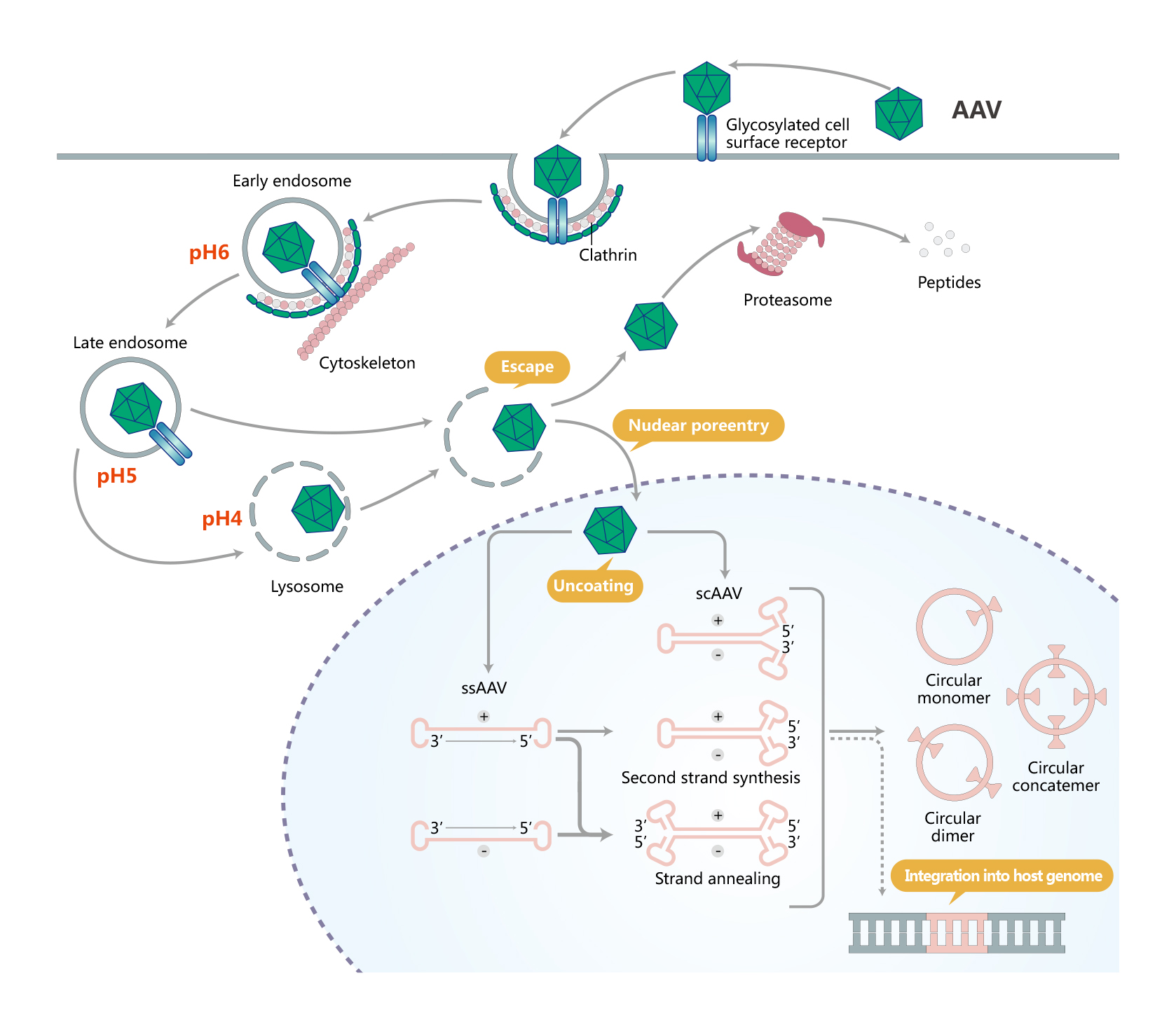
重组AAV载体可以通过将内源性的rep和cap基因替换为一个表达盒,该表达盒由一个启动子驱动一个感兴趣的转基因和一个poly(A)尾巴组成。通过包装质粒反式提供rep和cap基因以及腺病毒辅助质粒来包装载体。病毒编码序列的完全去除使AAV的包装能力最大化(包装容量为4.5Kb),并且有助于它们在体内递送时的低免疫原性和细胞毒性。
基因工程化的AAV能转导外源基因,借助特定启动子、光遗传学/化学遗传学元件、钙指示剂、神经递质探针或Cre/lox P,Flp/FRT重组酶技术,可选择性地标记特定的神经元。重组AAV基因传递效果好、缺乏致病性和安全性高、宿主细胞范围广、在体内表达时间长,是目前最有前途和最成功的基因治疗载体之一。
现阶段研究人员已发现12种人类AAV 血清型(AAV1至AAV12)和 100多种非人类灵长动物AAV血清型。不同AAV血清型具有不同的衣壳蛋白空间结构、序列和组织特异性,因而其识别与结合的细胞表面受体也相应有很大差别,这也导致不同血清型转染的组织类型、细胞类型和感染效率也各不相同。
已批准用于商业用途的:AAV1和AAV2;
临床使用频率最高的:AAV2;AAV8,AAV9,AAVRh10;
AAV8,AAV9:靶向全身的多种肌肉类型;
几乎所有天然AAV衣壳都可以在全身给药后有效地转到肝脏;
| Serotype | Glycan recognitiona | Coreceptor |
|---|---|---|
| AAV1 | Neu5Aca2-3GalNAcβ1-4GlcNAc | Unknown |
| AAV2 | 6-O-and N-sulfated heparin |
Fibroblast / hepatocyte growth factor receptor; laminin receptor; integrin αVβ5 and α5β1 |
| AAV3 | 2-O-and N-sulfated heparin | Hepatocyte growth factor receptor; Laminin receptor |
| AAV4 | Galβ1-4GlcNAcβ1-2Manα1-6Manβ1-4GlcNAcβ1-4GlcNAc | Unknown |
| AAV5 | Neu5Acα2-3(6S)Galβ1-4GlcNAc | Platelet-derived growth factor receptor |
| AAV6 | Neu5Acα2-3GalNAcβ1-4GlcNAc; N-sulfated heparin | Epidermal growth factor recepto |
| AAV7 | Unknown | Unknown |
| AAV8 | Unknown | Laminin receptor |
| AAV9 | Galactose | Laminin receptor |
| Tissue | Optimal Serotype |
|---|---|
| CNS | AAV1,AAV2,AAV4,AAV5,AAV8,AAV9 |
| Heart |
AAV1,AAV8,AAV9 |
| Kidney | AAV2 |
| Liver | AAV7,AAV8,AAV9 |
| Lung | AAV4,AAV5,AAV6,AAV9 |
| Pancreas | AAV8 |
| Photoreceptor Cells | AAV2,AAV5,AAV8 |
| RPE(Retinal Pigment Epithelium) | AAV1,AAV2,AAV4,AAV5,AAV8 |
| Skeletal Muscle | AAV1,AAV6,AAV7,AAV8,AAV9 |
1. 局部注射,直接在想表达的地方注射 rAAV ,适合器官水平特异感染,容易操作。
2. 使用组织/器官特异启动子来表达外源基因包装 rAAV, rAAV 即使感染了其他组织也由于特异性启动子而不会在其他组织中表达。
3.Cre-on,Flex,DIO,double floxed :使用 Cre-Loxp 系统,经过两次染色体重组在特定脏器中表达外源基因。
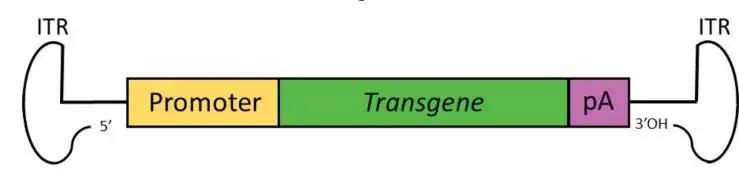
目前,研究人员常用的两类AAV分别为single-stranded AAV (ssAAV) and self-complementary AAV (scAAV)。scAAV是基于2个基础被开发的:一个是理论基础:无论scAAV还是rAAV,病毒颗粒中均可包裹二倍体,甚至四倍体的AAV基因组DNA;一个是结构基础:wtAAV ITR序列的特殊性(T型结构)。ssAAV包装基因组正义链和反义链的几率一样。ssAAV在入核、脱衣壳后,需要借助宿主DNA聚合酶或者分子间退火完成双链转化,才能启动基因转录过程,而scAAV中已存在双链,它入核后即可启动基因转录,跨过了双链转化的步骤,从而实现外源基因的快速表达。
ssAAV和scAAV向靶细胞中递送外源基因的过程:
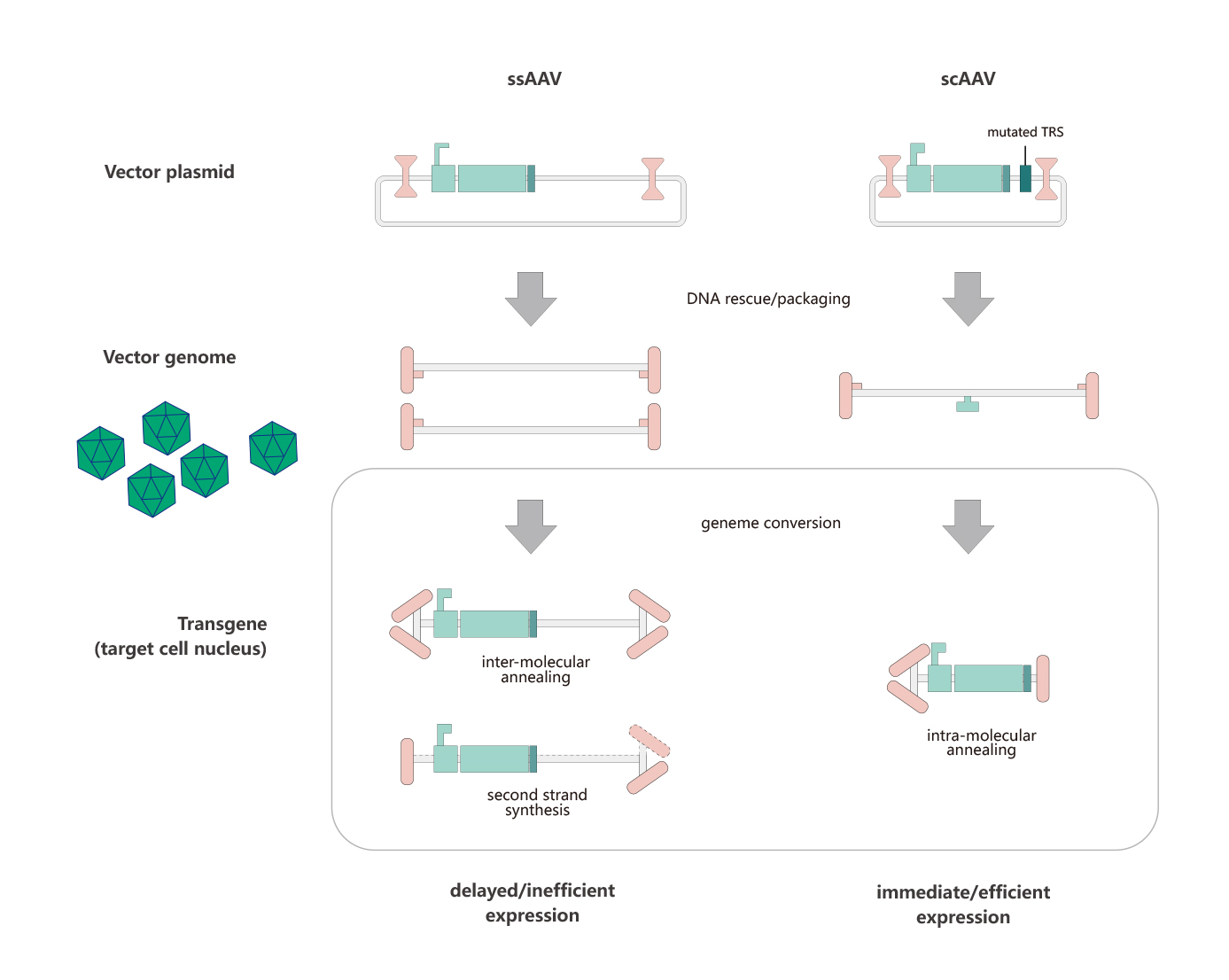
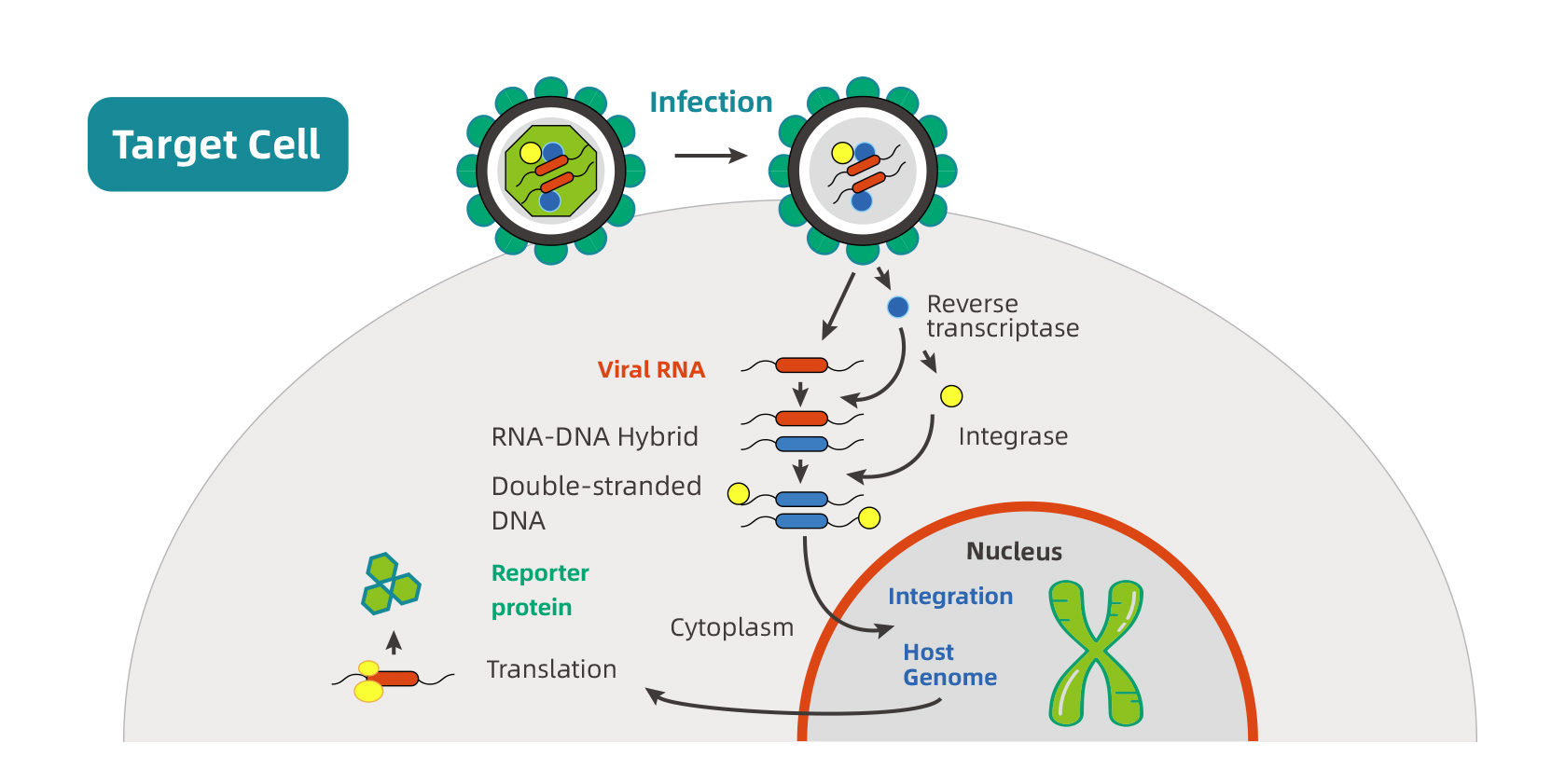
慢病毒属于逆转录病毒科,能够感染分裂和非分裂的细胞,从而将它们与普通的逆转录病毒区别开来。慢病毒基因组由两个拷贝的单链RNA组成,并被结构蛋白和酶蛋白包裹在衣壳中,这些结构蛋白和酶蛋白包括逆转录酶(将RNA转化为dsDNA)和DNA整合酶(将dsDNA整合到宿主基因组中)。
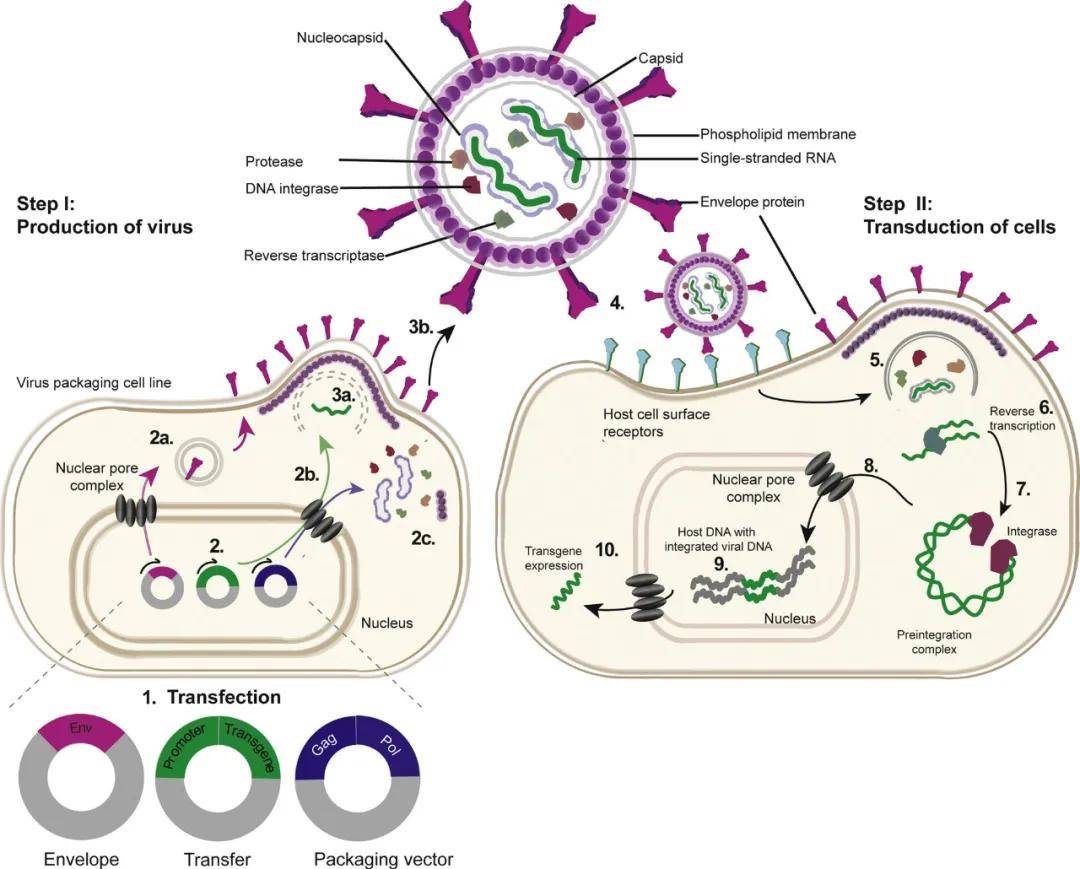
如上图步骤|,用包膜、转移和包装载体转染慢病毒包装细胞系。包膜、转移和包装载体转录的mRNA分别被翻译成:通过内质网进入细胞膜的病毒包膜蛋白(2a);单链RNA病毒基因组(2b);病毒结构蛋白和酶(2c),这三种成分组装成病毒颗粒,从宿主细胞膜上出芽。
如上图步骤‖,病毒颗粒通过包膜蛋白附着在宿主细胞表面受体上,病毒与宿主质膜融合释放结构蛋白、酶蛋白和病毒基因组。病毒RNA被逆转录成双链DNA,并与整合酶形成预整合复合物,整合酶通过核孔复合物,催化病毒DNA整合到宿主基因组中,最后转移载体启动子驱动转基因表达。
人类免疫缺陷病毒-1型(HIV-1)作为典型的慢病毒,其基因Gag、Pol和Env分别编码结构核心蛋白、逆转录酶和整合酶以及包膜糖蛋白;Tat和Rev参与病毒复制:Tat启动病毒基因组转录,并受长末端重复序列驱动,Rev促进病毒mRNA的核输出,并受到Rev响应元件的调控;包装信号(Ψ)是将病毒基因组进入衣壳的关键。此外,HIV-1基因组中包含的毒力因子Vif、Vpr、Vpu和Nef会干扰宿主防御机制,并包含在组装好的病毒粒子中。(图2)

慢病毒是以HIV-1为基础发展起来的基因治疗载体。
第一代慢病毒载体将所有包装元件编码在一个载体上,生物安全风险较高;
第二代慢病毒载体去除了毒力因子并将包膜从包装载体中分离,提高了安全性并增强了病毒复制,而不干扰有功能的病毒粒子的生产。此外,3’LTR包含U3缺失,可以消除病毒基因组复制的启动子活性,并在不影响病毒粒子滴度或转基因表达的情况下自灭活载体。
第三代慢病毒载体从包装载体中去除Rev,并用转移载体上游的一个组成启动子替换Tat,是目前被广泛应用的慢病毒包装载体。(图3)

引物是指在PCR反应中与待扩增的靶DNA区段两翼互补的寡聚核苷酸,其本质是单链 DNA片段。PCR扩增产物的大小及扩增的靶序列在基因组中的位置是由引物限定的,因此,引物的选择对PCR 成功与否具有决定性意义。引物设计质量是影响PCR扩增成败的关键因素,一般要考虑以下几个问题:
① 引物长度一般16~30 bp为宜、20~24 bp为佳。引物长度较短一般会降低扩增特异性,但会提高有效性,扩增的产物种类会增多。
② G+C含量一般为40%~60%为佳,两条引物G+C含量应尽量相似,Tm最好接近,差异最好在1℃以内,最多不超过5℃。
③ 两个引物之间不应发生互补,特别应避免形成"引物二聚体",如果无法避免,其3'端互补不应大于2个碱基,应尽量避免数个嘌呤或嘧啶连续排列,避免同源序列(尤其 6个碱基以上相同)。
④ 引物内部避免形成次级结构,尤其发卡结构,若用人工判断,引物自身连续互补碱基不能大于3bp,必要时应通过计算机进行结构分析。
⑤ 引物的延伸从3'端开始,3'端不能进行任何修饰,也不能有形成任何二级结构的可能,3'末端最后1个碱基最好是G或C。
⑥ 引物的5'端限定着PCR产物的长度,对扩增特异性影响不大,因此,5'端可以被修饰而不影响扩增的特异性。
⑦ 引物3'端不要终止于编码区城密码子的第3位,因密码子的第3位易发生简并,会影响扩增特异性与效率。
引物设计可借助于一些计算机引物设计软件方便地进行。目前用于引物设计的钦件有多种,其中Oligo6、Prmer5.0是非常方便且功能强大的引物设计分析软件。
聚合酶链式反应(polymerase chain reaction,PCR)是 Kary M u l l i s 于1985年发明的核酸体外扩增技术,是分子克隆研究中不可或缺的基本技术。随着分子生物学的发展,人们在PCR基本技术的基础上,不断地推陈出新,发展了多种PCR延伸技术(反转录PCR、锚定PCR、巢式 PCR、实时定量PCR等等),以适应不同的用途。
在有DNA单链、与DNA单链互补的寡核苷酸引物、dNTPs及DNA聚合酶的情况下,引物与单链的互补区结合后,在DNA聚合酶的催化作用下即可进行DNA单链的5'→3'合成反应。通过特殊的仪器不断满足上述条件,则DNA单链的5'→3'合成反应可不断进行下去,直到条件不复存在,实质上是模拟天然DNA的复制过程。
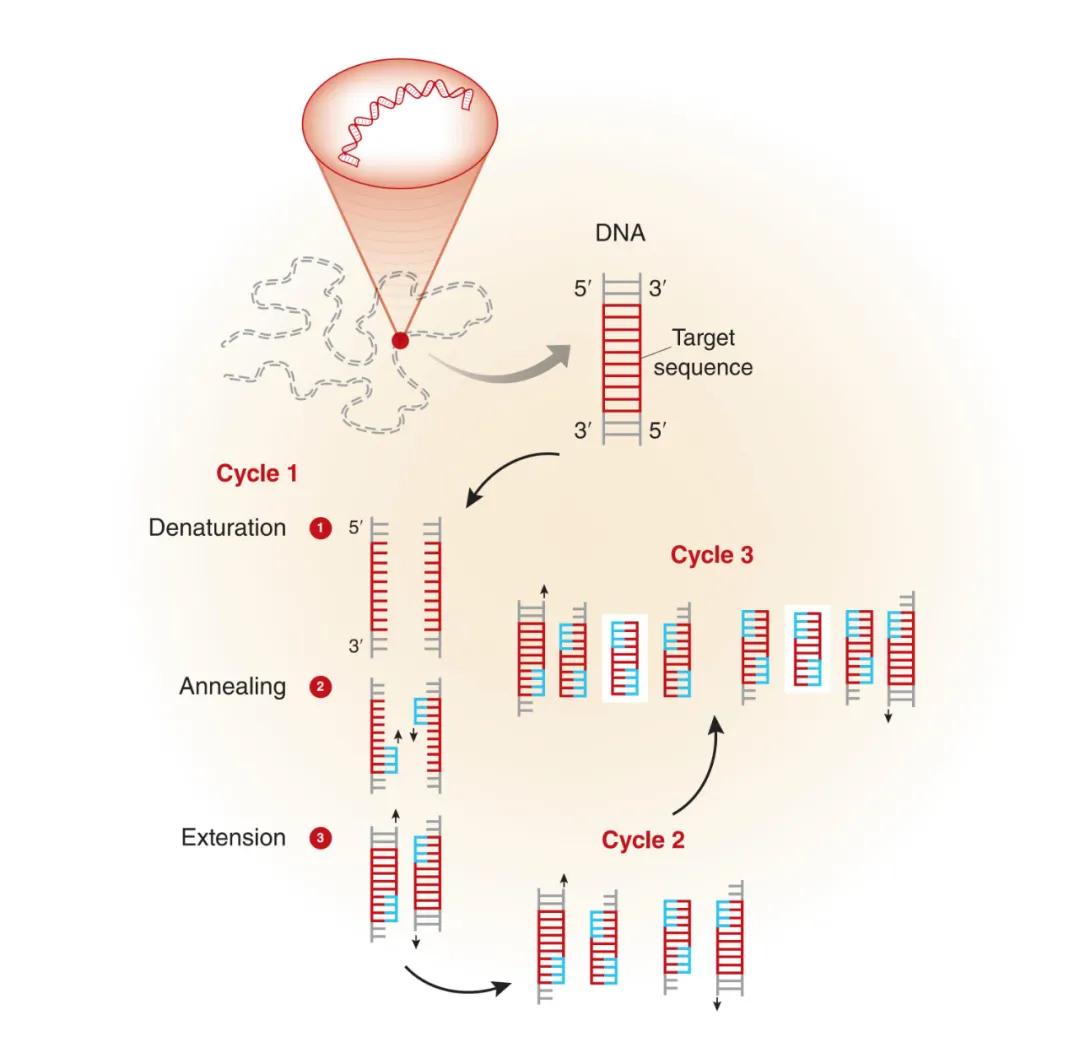
PCR包括三个基本过程:变性、退火和延伸。
①变性(denaturation):92~96℃的高温处理可使模板DNA双链间氢键断裂,解离成单链但不改变其化学性质,变性的时间一般为30 s,如果模板的G+C含量较高,变性时间可适当延长。
②退火(annealing):将温度降低到55℃左右,使引物与模板的互补区结合。退火的温度取决于引物的Tm值,并通过预备试验最终确定。一般引物越短,其Tm值也越低,反之则高。退火温度越高,引物与模板结合的特异性增强,非特异性的扩增概率降低,但扩增效率也随之降低。相反,随着温度的降低,引物与模板的非特异性结合的概率提高。退火的时间一般为30s。
③延伸(extension):在72℃温度下,DNA聚合酶将dNTPs连续加到引物的3'-OH端,以互补的DNA单链为模板,沿着模板延伸合成模板 DNA的互补链,称为延伸。延伸的时间由扩增产物的大小决定,一般每1kb延伸1min。
这三个过程组成一个循环周期,每个周期合成的产物又可作为下一个周期的模板,如此循环往复,经过n轮循环后,靶DNA的拷贝数理论上呈2n增长。
PCR反应经过一定数量的循环后,随着产物的对数累积趋于饱和,DNA片段不再呈指数积累,而是进入线性增长期或静止期,此过程称为平台效应(plateau effect)。
平台期与下列因素有关:①随着引物、dNTP 快速掺入底物中,它们的浓度降低,掺入速度减慢;②随产物增加,酶与模板的比例下降;③非特异性产物或引物二聚体与反应物的竞争;④产物的再结合。
PCR反应需要一定的条件才能完成,这些条件主要有以下方面:
(一)缓冲液
任何一个生化反应都必须在一定的缓冲体系中进行,缓冲体系除了提供一定的pH缓冲能力外,还有一些有助于反应进行的成分。PCR反应缓冲液通常采用10 mmol/L Tris-HCl(pH 8.3-9.0,室温)缓冲液体系,其中含有可激活DNA聚合酶的活性中心的二价阳离子,一般采用Mg2+,以MgCl2的形式提供,Mg2+的浓度高低可显著影响 PCR扩增产物的特异性,此外,缓冲液中还含有50 mmol/L的K+,有利于引物与模板的退火。有些缓冲液中还加入明胶或血清白蛋白(100 μg/mL)及去污剂(如Twcen20 等),对Taq DNA聚合酶起稳定作用。
(二)模板
PCR模板是含有待扩增序列的 DNA、mRNA或cDNA等,模板数量可直接影响扩增的效果。对于一般的 PCR扩增,104~107个模板分子可达到满意的效果,一般宜用纳克(ng)级的克隆DNA、微克(μg)水平的染色体DNA或102~105拷贝待扩增的DNA片段作为起始材料。除了数量外,模板的质量也非常重要,不能有太多的蛋白质和其他污染,特别是其他DNA的污染,即使是痕量的DNA,也会导致非特异性产物。
(三)dNTP
dNTP是dATP、dTTP、dGTP、dCTP的总称。贮备液为pH 7.0 左右,其浓度一般为20 mmol,-20℃保存。体系中为20~200 μmol即可,过低则反应速度下降,浓度过高则特异性下降,因为dNTP能络合溶液中的Mg2+,产生错配或抑制Taq DNA聚合酶活性。
(四)耐热的DNA聚合酶
PCR反应中使用的 DNA聚合酶必须耐高温,在90℃以上的高温下仍能有活性,正是由于耐热DNA聚合酶的应用才使 PCR技术得以实现,目前使用的耐高温DNA聚合酶主要有Taq DNA聚合酶、Tth DNA聚合酶、Vent DNA聚合酶、Pfu DNA聚合酶和Pwo DNA聚合酶等。
(五)引物
根据大量的研究报道,PCR反应的最佳条件为:①Taq DNA聚合酶的浓度为1.0-2.5 U/100μL反应液;②dNTP的浓度为20~200 μmol/L;③Mg2+浓度为0.5~2.5 mmol/L;④引物的浓度为0.2~1.0 μmol/L。在准备具体的PCR反应时,还要针对模板特性、PCR仪等进行上述反应体系的优化,并设置试验确定连退火温度、延伸时间,建立其相应的PCR反应最优程序。
在PCR过程中,研究人员通常会在PCR反应体系中添加二甲基亚砜DMSO,那么它究竟有什么作用呢?机理是什么呢?
通常我们可以这么理解:DMSO主要用于高GC含量的模板扩增,可能的机理是改善GC含量高的DNA的变形情况,降低其二级结构,使得聚合酶在二级结构处延伸。DMSO可以提高PCR的特异性,帮助扩增一些难扩的模板。当体系中加入DMSO时,可适当降低退火温度。
延伸阅读
在实践中,聚合酶链式反应可能因各种原因而失败,通常表现为两个问题,一是目的模板的扩增量太少,二是非目的基因扩增太多。这种情况话,研究人员通常会在反应体系中加入合适的添加剂进行解决。通常添加剂的作用主要是降低基因的二级结构或者降低非特异性的启动。下面是几种常用的添加剂以及它们的作用:
① 二甲基亚砜DMSO、非离子性洗涤剂、甜菜碱——可以降低DNA二级结构
② 甲酰胺、四甲基氯化铵TMAC——降低非特异性启动
③ 镁离子——聚合酶不可缺少的一个辅因子
④ 牛血清白蛋白——减少污染物
一般而言,常规PCR很难通过基因扩增产物来定量该基因在模板中的表达,而实时定量PCR(real-time quantitative PCR)则可解决这一问题。所谓实时定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对模板中特定基因进行定量分析的方法。
荧光扩增曲线可以分成三个阶段∶荧光背景信号阶段、荧光信号指数扩增阶段和平台期。在荧光背景信号阶段,扩增的荧光信号被荧光背景信号所掩盖,无法判断产物量的变化;在平台期,扩增产物已不再呈指数级增加,PCR产物量与起始模板量之间没有线性关系,无法根据最终PCR产物量算出起始DNA拷贝数。只有在荧光信号指数扩增阶段,PCR产物量的对数值与起始模板量之间存在线性关系,可以选择在这个阶段进行定量分析。
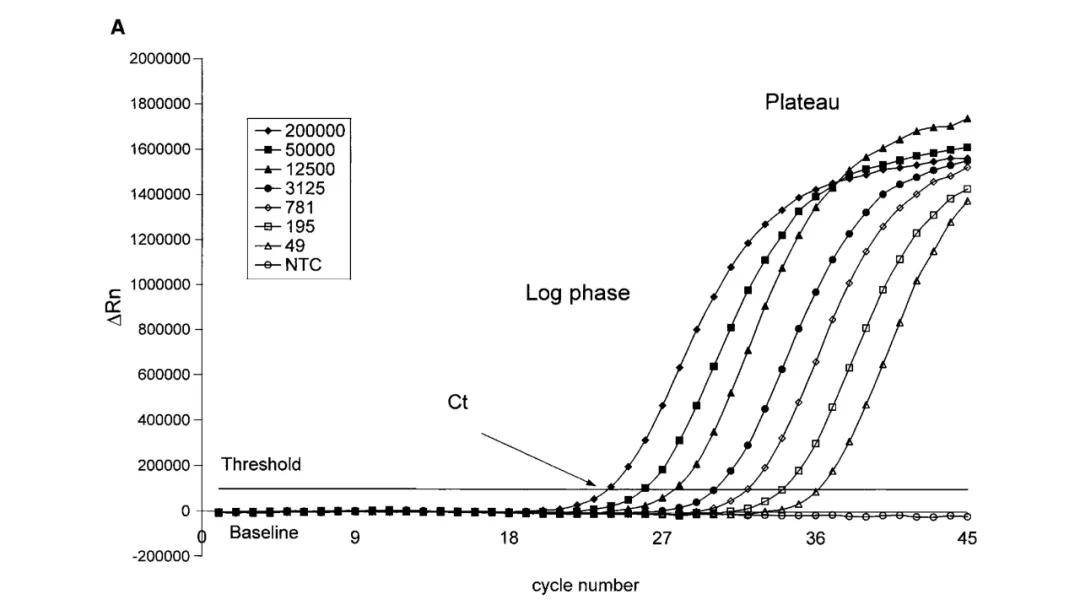
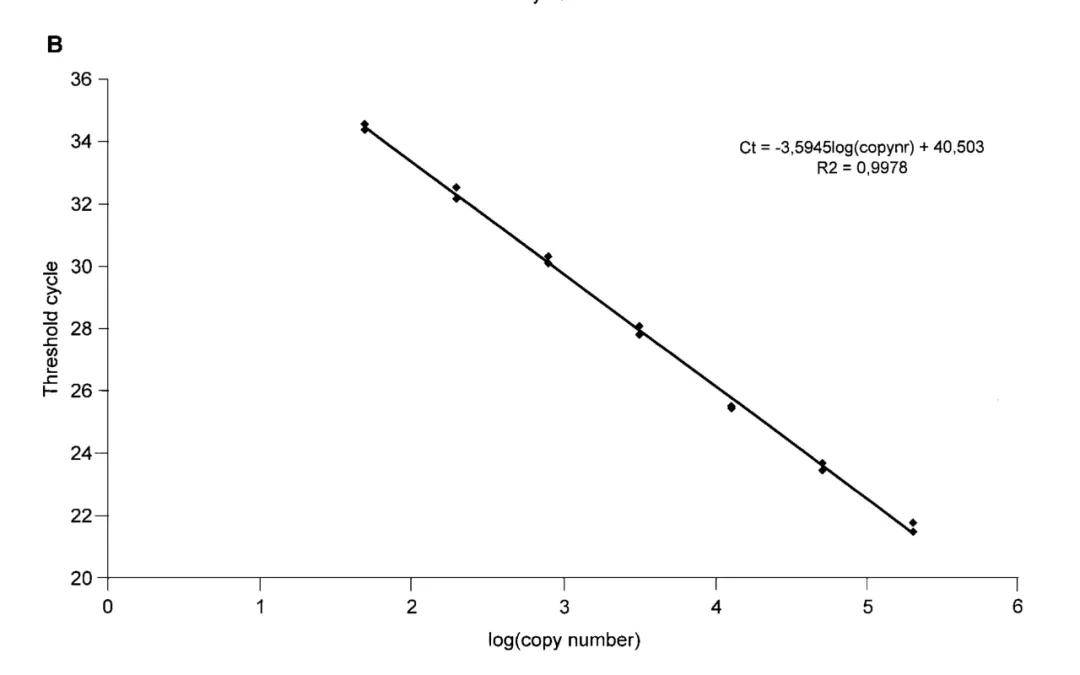
①临界点选择。临界点是在荧光扩增曲线上人为设定的一个值。它可以设定在荧光信号指数扩增阶段任意位置上,但一般将临界点缺省设置为3-15个循环的荧光信号的标准偏差的10倍;②临界点循环数(threshold cycle,Tc或Ct),PCR反应管内的荧光信号到达设定的阈值时所经历的循环数称为临界点循环数,模板的Ct值与模板的起始拷贝数的对数存在线性关系,起始拷贝数越多,Ct值越小;③标准曲线绘制。利用已知起始拷贝数的标准品可绘制标准由线,其中横坐标代表起始拷贝数的对数,纵坐标代表Ct值。因此,只要获得未知样品的Ct值,即可从标准曲线上推算出该样品的起始拷贝数。
探针类和非探针类实时定量PCR的化学原理有所不同。探针类是利用与靶序列特异杂交的探针来指示扩增产物的增加;非探针类则是利用荧光染料或者特殊设计的引物来指示扩增产物的增加。前者由于增加了探针的识别步骤,特异性更高,但后者则简便易行。
①TaqMan荧光探针 PCR扩增时在加入一对引物的同时,也加入一个特异性寡核苷酸荧光探针,其两端分别标记了一个报告荧光基团和一个淬灭荧光基团。在探针完整的情况下,报告基团发射的荧光信号会被淬灭基团吸收;PCR扩增时,Taq DNA酶的5'-3'核酸外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。
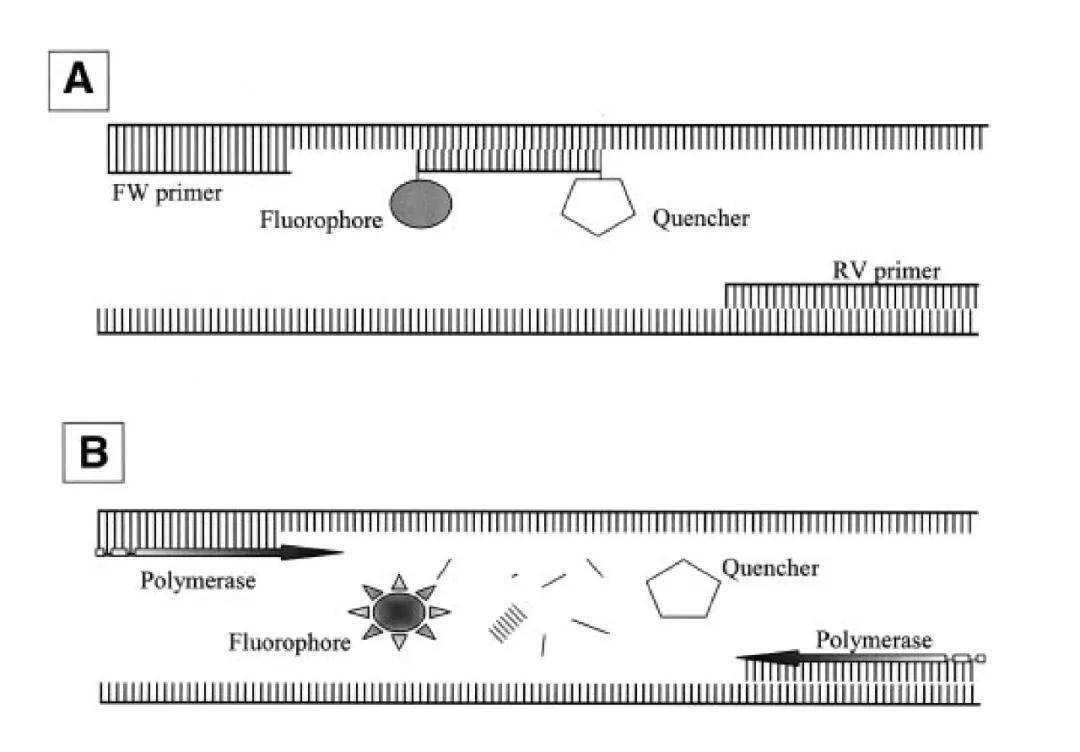
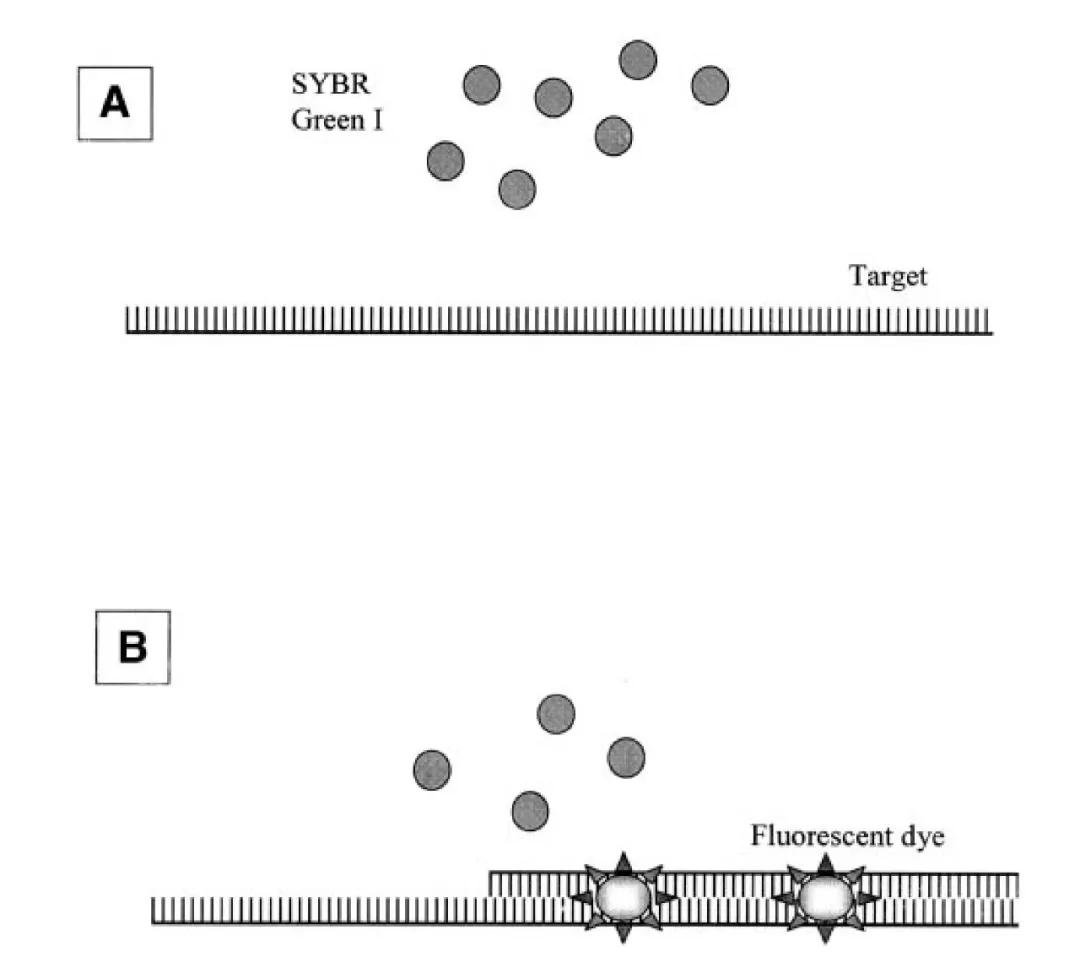
②SYBR荧光染料 在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料特异性地掺入DNA 双链后,发射荧光信号,而不掺入链中的SYBR 染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。
实时荧光定量PCR技术是DNA定量技术的一次飞跃,运用该技术可进行基因表达、基因型鉴定、DNA或RNA的绝对定量等领域的分析。

400-077-2566

service@wzbio.cn







