AAV在脑组织中的靶向策略(应用篇)
· 案例一 ·
“TNF-α-dependent neuronal necroptosis regulated in Alzheimer’s disease by coordination of RIPK1-p62 complex with autophagic UVRAG”
阿尔茨海默病(AD)是一种常见的神经退行性疾病,临床表现为认知功能障碍和记忆力减退等,其病理特征包括Aβ蛋白沉积、Tau蛋白过度磷酸化、神经炎症和神经元坏死等。研究表明,AD发病的病理过程可能与神经元坏死和自噬有关。神经元坏死是AD的主要特征,程序性细胞坏死与AD的发病机制相关,然而AD中诱导神经元坏死的机制尚不清楚。自噬受损导致Tau蛋白的积累,这是包括AD在内的大多数神经退行性疾病(NDs)的特征。然而,目前对AD发病机制中自噬途径和神经元坏死之间可能的相互作用知之甚少。TNF-α作为关键促炎因子,其诱导的炎症可能对神经元死亡产生不利影响,但在AD发病过程中TNF-α的升高是否能引起神经元坏死还尚未可知。
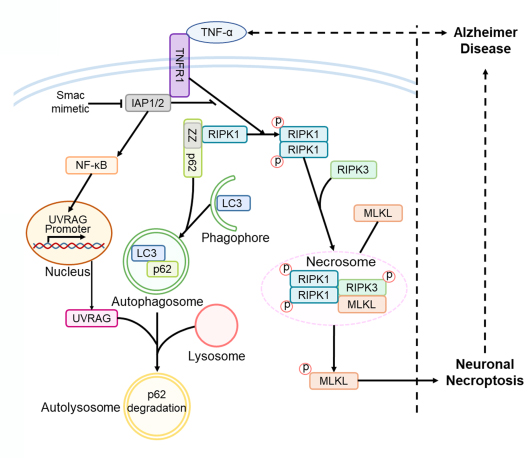
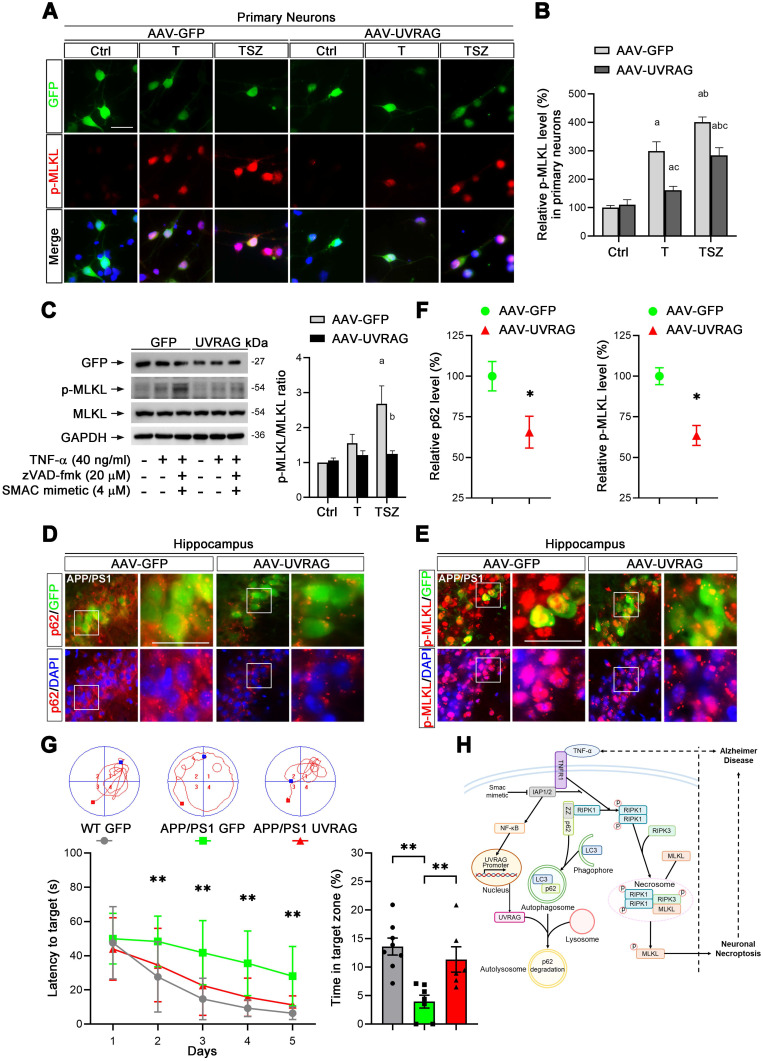
图1. 阿尔茨海默病中神经元坏死的调控机制
本研究利用来自AD患者的脑组织和AD小鼠模型,通过免疫组织化学 (IHC) 染色和免疫印迹法测定坏死信号通路中关键基因的表达,发现TNF-α/TNFR1信号通路在AD神经元坏死激活中发挥作用。在TNF-α刺激下,积累的p62 招募 RIPK1并诱导其自我寡聚,激活下游 RIPK1/RIPK3/MLKL级联反应,导致神经元坏死。进一步研究发现,p62的异常积累是由TNF-α诱导的UVRAG下调介导的自噬损伤引起的。通过注射AAV载体上调UVRAG的表达,可以抑制 AD细胞和小鼠模型中神经元坏死,并改善认知功能。本研究揭示了AD发病过程中神经元坏死和自噬机制之间的联系,为AD的临床干预提供了潜在的靶点,同时也为其他NDs的细胞死亡调节机制的探索提供了基础。
为探究AD患者脑内TNF-α水平的升高是否会导致神经元的坏死,作者检测了AD患者和小鼠的脑组织中MLKL和p-MLKL(坏死标志物)的表达,发现在AD中细胞坏死确实被激活。通过在小鼠侧脑室注射TNF-α,发现高水平的TNF-α可以增加p-MLKL的表达并激活坏死。使用AAV9对TNFR1(TNF-α受体)进行敲低,发现TNFR1敲低后能导致p-MLKL表达水平的降低,提示TNF-α/TNFR1信号通路在AD神经元坏死过程中发挥重要作用。
图2. TNF-α/TNFR1诱导神经元坏死
研究人员发现在TNF-α诱导下,p62积累并激活下游 RIPK1/RIPK3/MLKL级联反应,导致了神经元坏死,而p62的异常积累是由UVRAG下调介导的自噬流受损引起的。为进一步探究UVRAG在AD小鼠神经元坏死中的作用,研究者利用AAV载体在原代神经元中对UVRAG进行了过表达,结果发现UVRAG的过表达明显降低了TNF-α诱导的MLKL磷酸化。通过在AD小鼠海马区注射AAV-UVRAG,发现p62与MLKL的磷酸化水平均显著降低。此外,UVRAG的过表达明显改善了AD小鼠的学习和记忆功能障碍。以上结果表明,上调UVRAG可抑制AD小鼠神经元坏死并改善认知功能,提示UVRAG可能是AD干预的潜在靶点。
图3. UVRAG抑制原代神经元和AD小鼠中的神经元坏死
· 案例二 ·
“Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease”
帕金森病(PD)是常见的神经退行性疾病,已有研究表明,α-突触蛋白(α-syn)诱导的小胶质细胞活化是PD发病的重要因素之一,代谢型谷氨酸受体5(mGluR5)及其信号通路在保护神经元免受神经炎症中发挥重要作用,且mGluR5可能在α-SYN诱导的PD神经炎症和神经毒性的发病机制中起重要作用,但其具体调控机制尚不明确。为探讨mGluR5在α-SYN诱导的小胶质细胞炎症中的作用和机制,作者通过过表达α-SYN来激活小胶质细胞,发现mGluR5的激活部分抑制了α-SYN诱导的炎症反应,保护小胶质细胞免受神经毒性。此外,α-SYN通过与溶酶体结合来促进mGluR5的降解机制在PD动物模型中同样存在。这些发现为解密mGluR5调控α-SYN诱导的小胶质细胞炎症机制,探究PD未知的发病机制提供了新的思路。
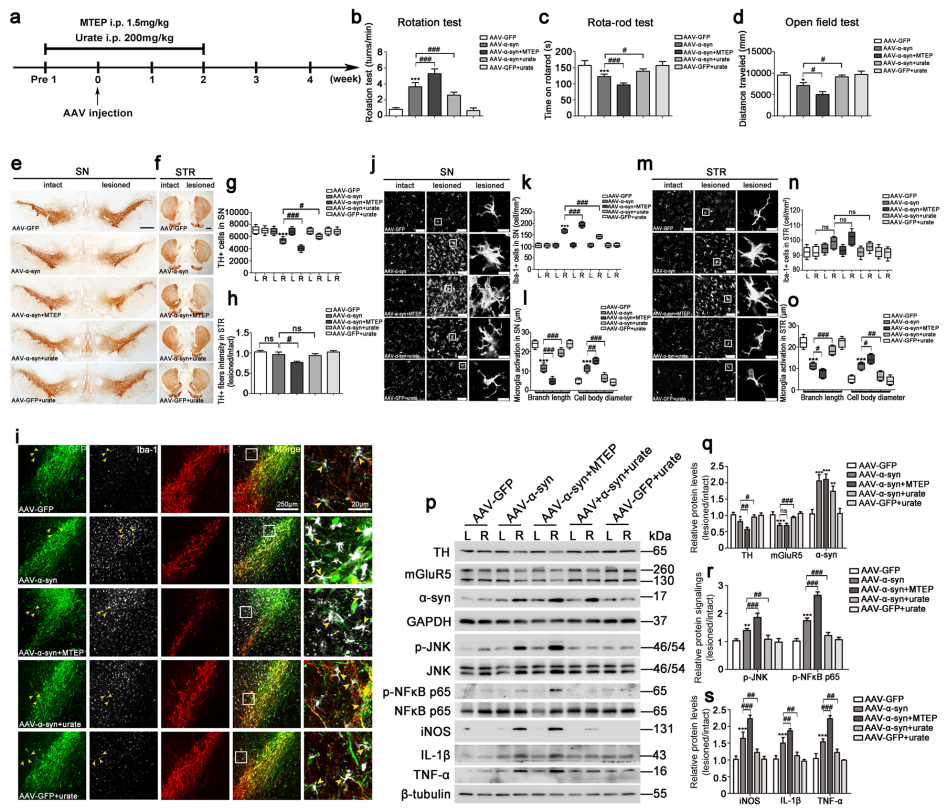
为确定mGluR5在α-syn诱导的体内小胶质细胞激活中的功能作用,作者利用AAV9-α-syn诱导PD大鼠模型,并对其进行行为评估及病理生理学分析,发现α-syn过表达组从第8周至第16周出现显著的进行性功能障碍,且过表达α-syn,能导致mGluR5表达降低,同时TH表达改变;此外小胶质细胞中的炎性因子iNOS、IL-1β和TNF-α的表达水平在注射后10天开始升高,4周时达到峰值,8周后影响消失,这说明在PD的发病早期伴随着反应性小胶质细胞的高度局部炎症反应。为进一步确定炎症干预在PD早期的作用,作者在第4周炎症高峰期时利用MEPT(mGluR5拮抗剂)或尿酸盐处理,结果发现MEPT处理阻断了mGluR5的活性并加剧了炎症反应,而尿盐酸处理减缓了炎症反应。综上,在AAV-α-syn诱导的PD模型中,mGluR5的激活在介导神经保护的抗炎反应中起着关键作用。
图4. mGluR5在AAV-α- sync诱导的大鼠PD模型中具有抗炎作用
· 案例三 ·
“PHLDA1 promotes microglia-mediated neuroinflammation via regulating K63-linked ubiquitination of TRAF6”
PD作为一种常见的年龄相关性神经退行性疾病,以黑质致密部(SNc)中多巴胺能(DA)神经元的进行性丢失为特征。小胶质细胞介导的神经炎症在PD等神经退行性疾病的进展中发挥着重要作用。目前普遍认为,TLR4/TRAF6/NF-κB或MAPKs信号通路在小胶质细胞活化介导的神经炎症的调节中发挥关键作用。已知PHLDA1(人Plecstrin同源域家族A成员 1,也称T细胞死亡相关基因51,TDAG51)参与了对TLR(Toll样受体)激活的免疫反应的调节,然而,PHLDA1在免疫应答中的确切分子信号和功能作用尚未完全阐明,特别是在小胶质细胞激活和神经退行性疾病中的功能作用尚不清楚。
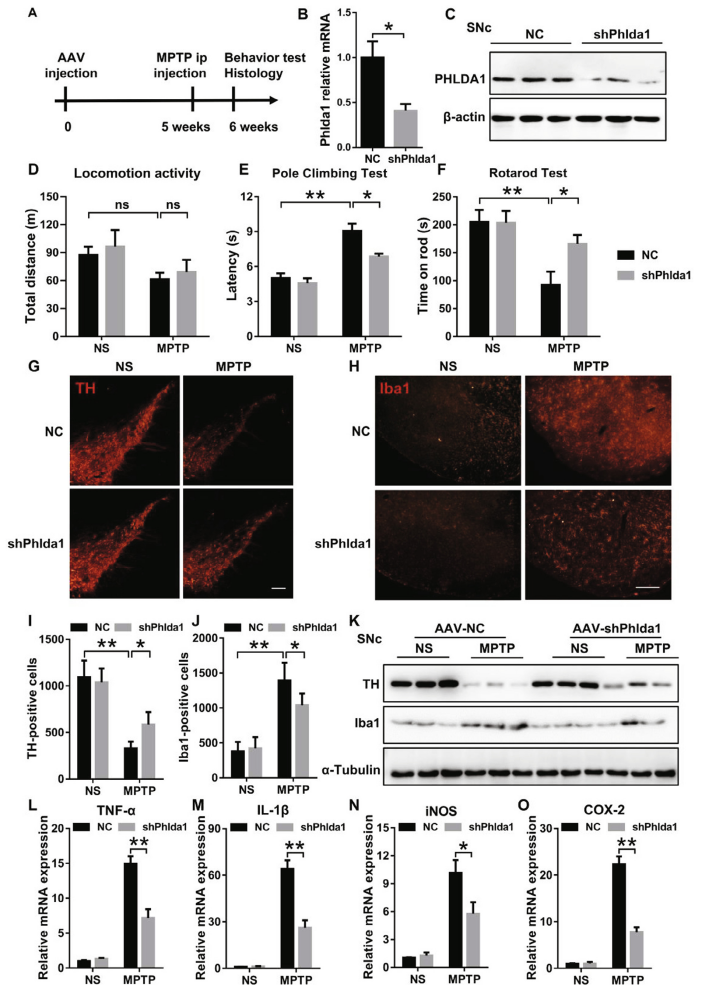
研究人员发现在体内和体外小胶质细胞中,PHLDA1的表达在炎症刺激下迅速增加。利用腺相关病毒(AAV)下调PHLDA1可改善MPTP诱导的小鼠运动缺陷并抑制神经炎症。在体内,发现LPS诱导的TNF-α、IL-1β、iNOS和COX-2等促炎基因表达在PHLDA1敲除的小胶质细胞中降低。机制研究表明,LPS刺激后,小胶质细胞中PHLDA1表达增加,导致与TRAF6的直接相互作用,并增强其K63连接的泛素化介导的NF-κB信号通路激活。PHLDA1缺乏可干扰TRAF6 k63的泛素化并抑制小胶质细胞炎症反应。本研究表明了PHLDA1与小胶质细胞介导的DA神经毒性有关,提示PHLDA1可能是一种有效的神经炎症调节剂,为治疗神经炎症相关疾病(如PD)提供新的药物靶点。
为探究PHLDA1在PD中的作用,研究人员利用AAV介导的shRNA对小鼠体内PHLDA1进行了敲降,5周后检测发现AAV9-shPhlda1组小鼠,PHLDA蛋白与mRNA水平均显著下降。感染小鼠给予MPTP诱导PD样表型,发现抑制PHLDA1的表达无法改善小鼠基础运动功能,但能显著减弱MPTP诱导的行为损伤,对ShPhlda1小鼠中脑的TH免疫组化染色和免疫印迹进一步证实了其保护作用,这表明抑制PHLDA1的表达可显著减轻MPTP所致的多巴胺能神经元和行为损伤。有趣的是,ShPhlda1小鼠SNc中Iba-1(小胶质细胞标志物)阳性细胞数量和总蛋白水平也降低,且ShPhlda1小鼠中炎性因子的表达降低。这些结果表明,SNc中PHLDA1的耗竭保护了DA神经元并抑制了神经炎症,最终减弱了MPTP诱导的PD样表型的发展。
图5. SNc中PHLDA1敲除可显著改善MPTP诱导的DA神经元损伤和运动障碍
· 案例四 ·
“Upregulation of KDM6B contributes to lipopolysaccharide-induced anxiety-like behavior via modulation of VGLL4 in mice ”
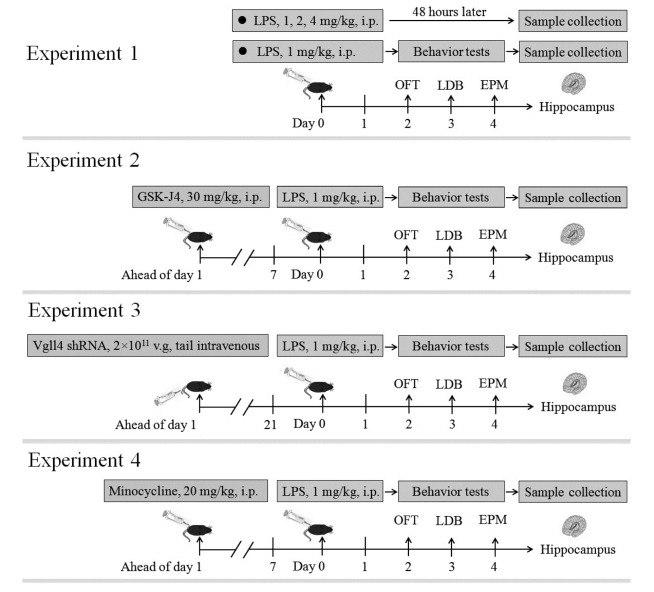
神经精神障碍如焦虑症,近年来已发展成为日益严重的公共心理健康问题。有证据表明,LPS诱导的神经炎症与焦虑样行为之间存在因果关系,阻断神经炎性介质已被认为是治疗焦虑的一种潜在方法。近年来,表观遗传学被认为在神经精神障碍如焦虑症中起着至关重要的作用,其中组蛋白H3K27me3去甲基化酶KDM6B作为一个关键的表观遗传调节因子,在炎症、细胞分化、神经再生、癌症以及中枢神经系统和行为反应等中发挥重要作用。然而,KDM6B在神经炎症诱发的焦虑样行为中的作用知之甚少。本研究探讨了KDM6B在LPS诱导的焦虑样行为中的潜在作用,并评估了其是否与VGLL4的调控有关。
首先,研究者使用LPS诱导小鼠焦虑样行为,并测定小鼠海马区和前额皮质中KDM6B和VGLL4的表达以及小鼠海马区焦虑样行为和神经炎症的变化,发现海马区KDM6B、VGLL4、IL-1β和Iba-1的表达水平呈LPS剂量依赖式增加,并伴随焦虑样行为的增加。接着,分析了KDM6B抑制剂GSK-J4对LPS诱导的焦虑样行为的改变,发现GSK-J4 处理减弱了LPS诱导的 VGLL4、STAT3、IL-1β 和 Iba-1的上调,并减轻了焦虑样行为。随后,用AAV介导的VgLL4 shRNA 敲低VGLL4,阻止了LPS诱导的小鼠海马区焦虑样行为和STAT3、IL-1β 和 Iba-1 表达水平的增加。最后,发现小胶质细胞抑制剂-米诺环素(MNC)可以减弱LPS诱导的焦虑样行为。本研究发现,LPS诱导的神经炎症促进了海马区中KDM6B 的激活,并且LPS诱导的焦虑样行为与海马区中KDM6B诱导VGLL4的上调有关。
图6. 研究思路
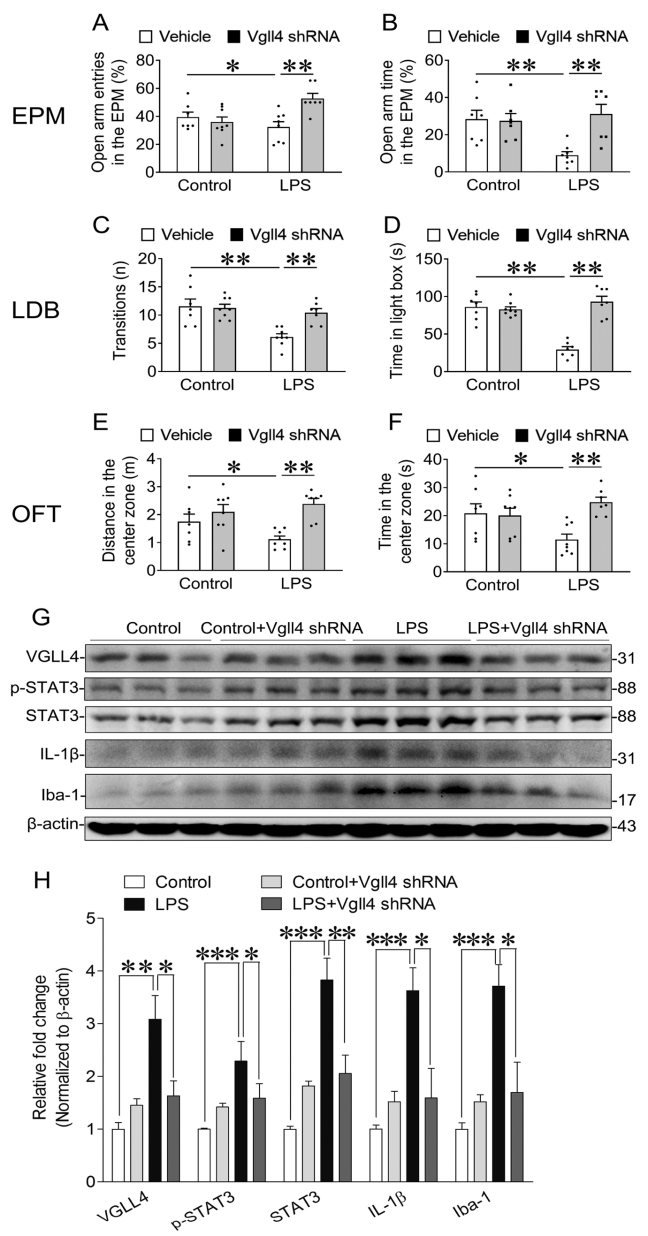
为进一步探究VGLL4在LPS诱导的焦虑样行为中的作用,作者在LPS注射前,使用AAV介导的VGLL4 shRNA对VGLL4进行敲低处理,通过旷场、迷宫等行为学测试发现,敲低VGLL4可以减轻LPS诱导的小鼠焦虑样行为。此外,LPS诱导了海马区STAT3(信号转导和转录激活因子3)表达的增加,而这种增加在敲低VGLL4后被逆转,并且VGLL4的敲低也显著减弱了LPS诱导的海马区IL-1β和Iba-1的表达。这些研究结果表明,VGLL4和海马中的STAT3之间存在促进关系,VGLL4通过上调海马中的STAT3来促进LPS诱导的焦虑样行为。
图7. VGLL4敲低对LPS诱导的焦虑样行为和海马中相关蛋白表达水平的影响


 当前位置:首页 > 研究领域 > 神经系统
当前位置:首页 > 研究领域 > 神经系统