

 当前位置:首页 > 研究领域 > 循环系统
当前位置:首页 > 研究领域 > 循环系统
临床证据表明,压力负荷引起的心脏肥大与不良心脏结局密切相关。内皮功能障碍是心脏肥大和心力衰竭(HF)进展的关键因素,富含亮氨酸重复序列8A(LRRC8A)是血管内皮稳态的关键调节因子,但其在压力负荷引起的病理性肥大和功能障碍中的功能作用仍不清楚。近期厦门大学附属心血管病医院张雁惠/揭领军/潘磊/李桂阳联合温州医科大学附属第二医院李磊团队在Angiogenesis (IF 9.2)上发表文章“Endothelial LRRC8A mitigates pressure overload-induced cardiac hypertrophy by promoting coronary angiogenesis”,发现内皮LRRC8A是压力负荷引起的肥厚心脏中冠状血管生成的关键调节因子,并表明它可以作为心脏肥大和心力衰竭的治疗靶点。
| 基因信息 | LRRC8A:富含亮氨酸重复序列8A |
|---|---|
| 病毒产品 | AAV9-ICAM2-NC;AAV9-ICAM2-LRRC8A |
| 实验动物 | 4周雄性C57BL/6小鼠 |
| 注射方式 | 尾静脉注射 |
| 注射剂量 | 1×1011 viruses,30μL |
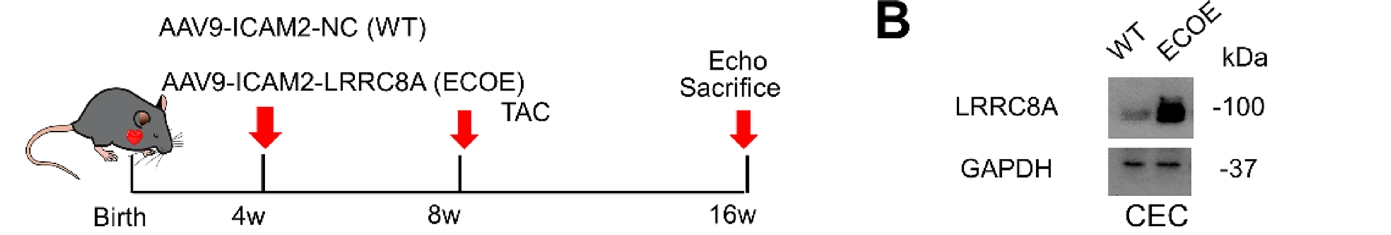
作者发现梗阻性肥厚型心肌病患者和接受TAC手术小鼠的心脏和心脏内皮细胞中LRRC8A下调,表明心脏内皮LRRC8A可能参与TAC驱动的病理性肥大发病机制。为了研究内皮LRRC8A缺陷对压力负荷诱导病理性肥大的直接影响,作者构建了内皮特异性LRRC8A缺失小鼠(ECKO),然后ECKO小鼠和WT小鼠接受TAC或假手术。超声心动图分析显示ECKO和WT假手术组之间的心脏结构和功能没有统计学显著差异,但与WT-TAC小鼠相比,ECKO-TAC小鼠的心功能较差,此外TAC-ECKO小鼠出现更严重的肥大。这些结果表明内皮LRRC8A缺陷会加剧病理性肥大和功能障碍。ScRNA-seq分析显示内皮LRRC8A在压力负荷引起的病理性肥大中充当血管生成基因。
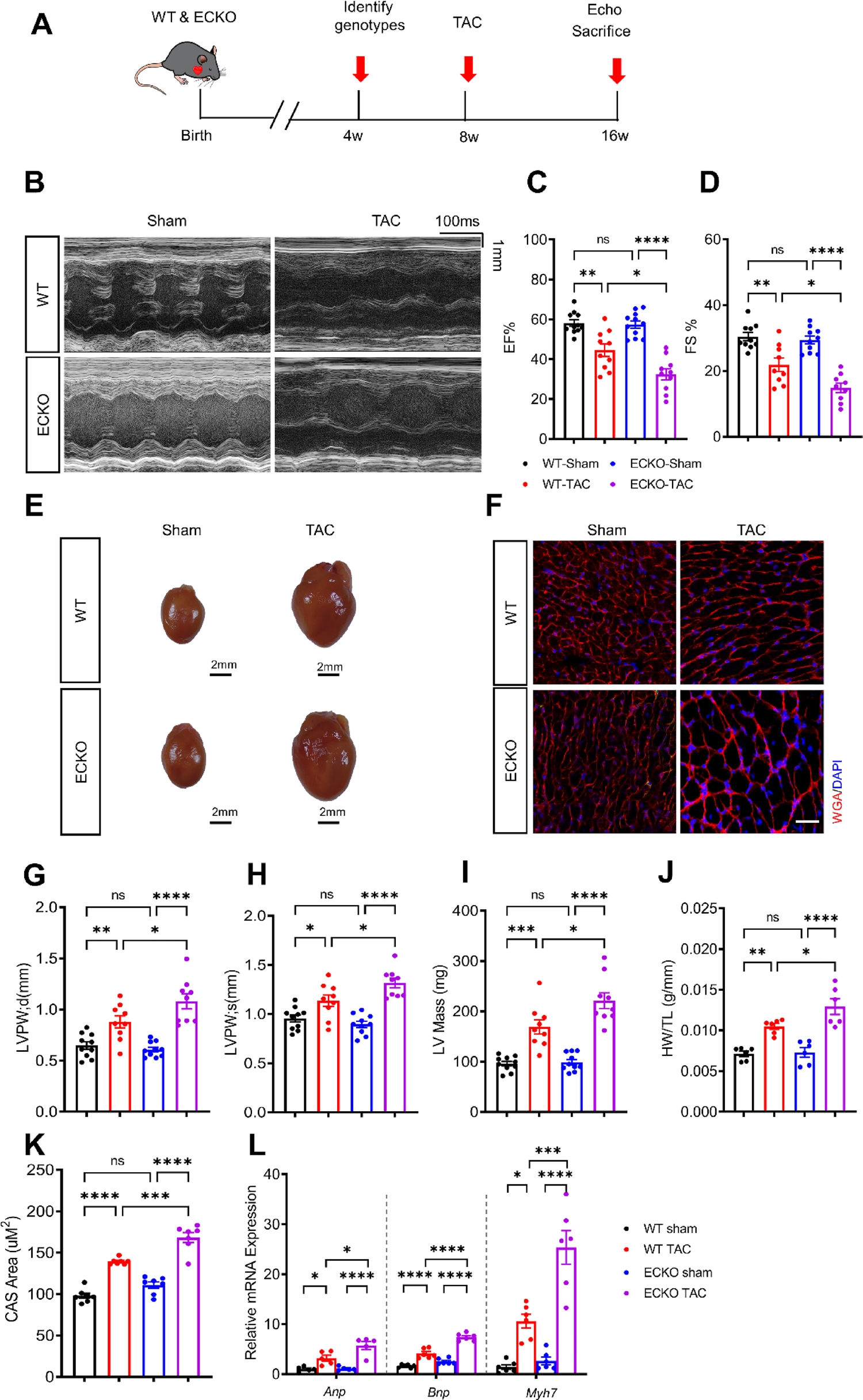
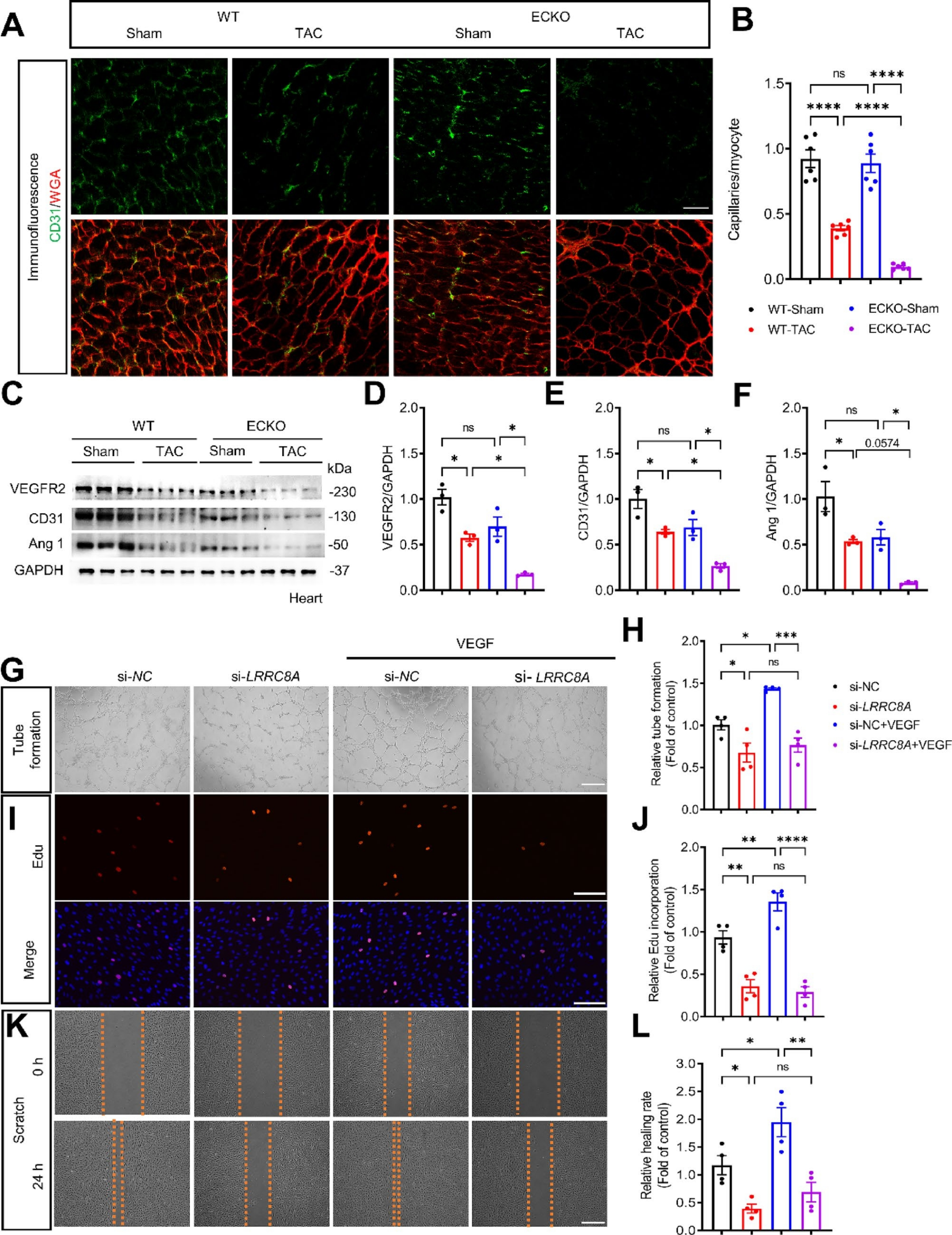
作者探究了内皮LRRC8A缺失对体内和体外血管生成的影响。发现TAC-ECKO小鼠的毛细血管密度低于TAC-WT小鼠,免疫印迹分析表明TAC-WT小鼠心脏中血管生成标记物(VEGFR2、CD31和Ang 1)表达下降,但TAC-ECKO 鼠中血管生成标记物表达进一步下降。作者还评估了LRRC8A是否影响管形成、迁移和增殖,使用siRNA敲低人脐静脉内皮细胞(HUVEC)中的LRRC8A,发现LRRC8A敲低明显减少了管形成,且LRRC8A缺陷型HUVEC的Edu阳性细胞比例低于对照组。伤口愈合测试表明LRRC8A缺陷的HUVEC的迁移能力显著下降。这些发现共同表明LRRC8A对于维持EC的VEGF响应功能至关重要,LRRC8A缺失显著抑制VEGF的促血管生成作用。机制研究表明LRRC8A调节VEGF-VEGFR2轴,并通过促进VEGF诱导的VEGFR2内吞来减轻压力负荷引起的心脏肥大和功能障碍。
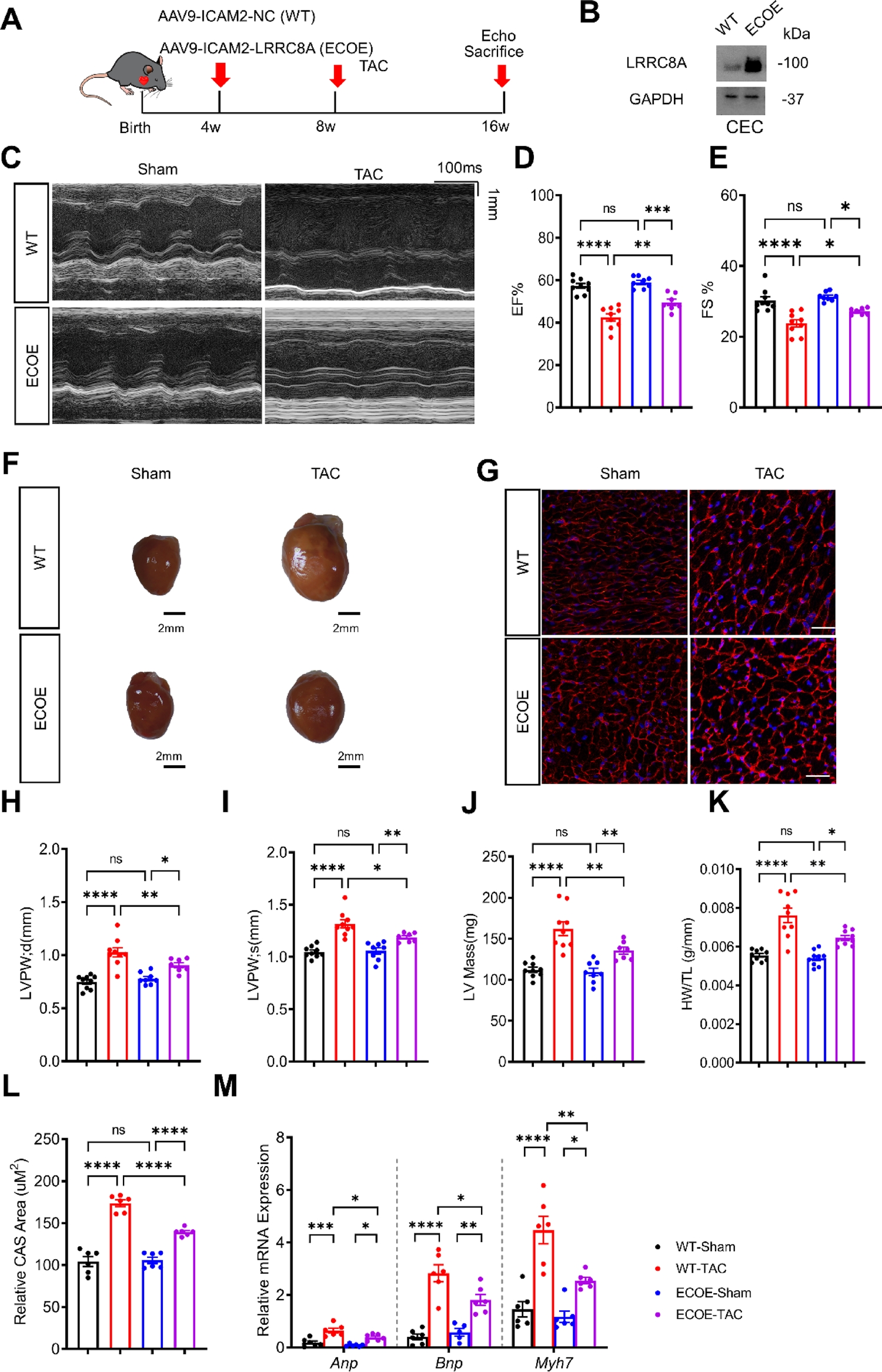
作者探究了LRRC8A过表达能否预防TAC诱导的病理性肥大和功能障碍,使用AAV9-ICAM2-LRRC8A生成内皮细胞特异性表达LRRC8A小鼠(ECOE),然后对ECOE和WT小鼠进行TAC手术,发现与WT-TAC小鼠相比,ECOE-TAC组的EF%和FS%得到改善,从而恢复了TAC诱导的心脏功能障碍。此外内皮LRRC8A过表达减弱了TAC诱导的病理性肥大,表现为HW/TL、LWPW、LWPWd、LV质量和心肌细胞横截面积减少,还明显降低了TAC诱导的Anp、Bnp和Myh7基因表达。作者进一步研究了内皮LRRC8A过表达对病理性肥厚心脏毛细血管密度的影响,发现与WT-TAC相比,ECOE-TAC心脏的毛细血管密度上调,血管生成标记物(包括VEGFR2和CD31)水平升高。这些结果表明AAV介导的LRRC8A过表达通过促进血管生成来恢复TAC诱导的病理性肥大和功能障碍。
本研究发现内皮LRRC8A是一种促血管生成蛋白,可通过正向调节VEGF-VEGFR2轴,促进VEGF诱导的VEGFR2内吞来减轻压力负荷引起的心脏肥大和功能障碍。这些发现确立了LRRC8A作为压力负荷响应性血管生成调节剂的生物学作用,也为心力衰竭治疗的前瞻性治疗提供了新的见解。

400-077-2566

service@wzbio.cn








