

 当前位置:首页 > 研究领域 > 循环系统
当前位置:首页 > 研究领域 > 循环系统
支链氨基酸在心脏生理学和疾病中发挥着关键作用,缬氨酸分解代谢酶ACAD8的遗传缺陷在临床上与人类异丁酰辅酶A失调和心肌病相关,但其在心脏病中的作用仍不清楚。近期中国医学科学院北京协和医学院刘德培院士、陈厚早教授团队联合四川大学华西第二医院唐小强教授团队在Nature Communications(IF 15.7)上发表文章“ACAD8 deficiency promotes pathological cardiac hypertrophy in response to pressure overload by regulating histone isobutyrylation”,阐明了ACAD8缺陷在心脏病中的作用,并揭示了转录调控和心脏病理学中的组蛋白异丁酰化。

| 基因信息 | ACAD8:酰基辅酶A脱氢酶家族成员8 |
|---|---|
| 实验动物 | 9周龄雄性C57BL/6小鼠 |
| 病毒产品 | AAV9-cTnT-ACAD8、AAV9-cTnT-Ctrl |
| 注射方式 | 尾静脉注射 |
| 注射量 | 0.8×1012 vg,100μl of viral dilution |
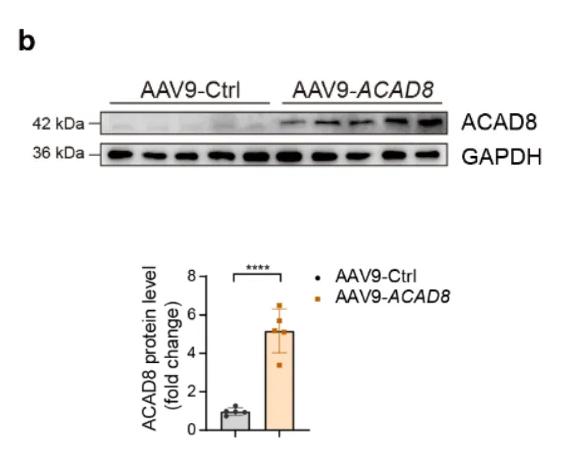
作者发现病理性心脏肥大中发生缬氨酸分解代谢受损和ACAD8缺乏,表明其可能在心脏肥大中发挥关键作用。为了研究缬氨酸分解代谢和ACAD8在病理性心脏肥大中的作用,作者构建了心肌细胞特异性Acad8敲除小鼠(Acad8cKO),并通过TAC手术诱导小鼠病理性心脏肥大。发现与对照小鼠相比,Acad8cKO小鼠表现出心脏功能障碍加剧,此外心脏重量与体重(HW/BW)和心脏重量与胫骨长度(HW/TL)的比率增加,同时心脏的横截面积和心肌细胞增大,心脏纤维化增加,以及肥大标记基因(Nppa和Nppb)表达增加。心脏肥大和心力衰竭进展常常伴随着细胞死亡,作者还使用TUNEL测定评估了心脏组织的细胞凋亡,发现Acad8cKO小鼠表现出更严重的细胞死亡。这些发现表明ACAD8缺陷直接促进病理性心脏肥大。
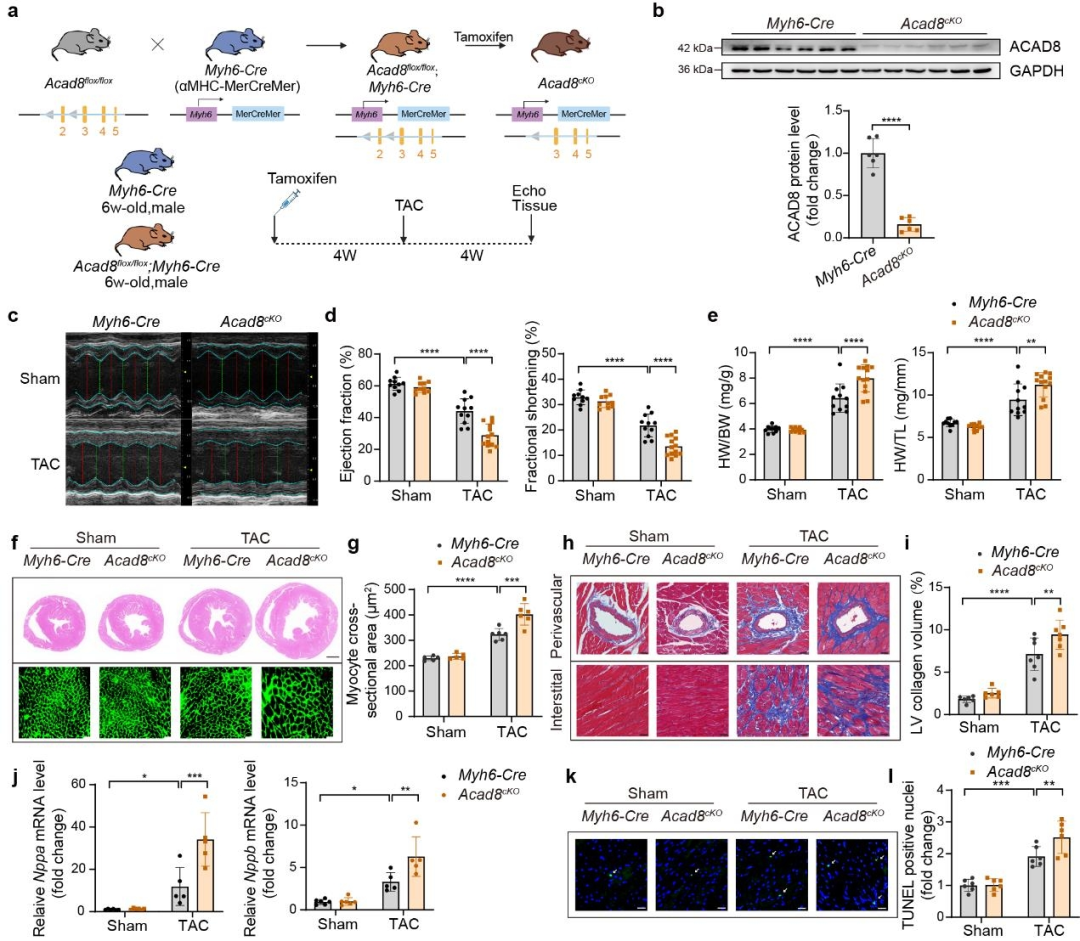
作者发现TAC手术心脏中ACAD8缺陷导致异丁酰辅酶A显著积累并促进组蛋白异丁酰化。对isobutyrate(异丁酰辅酶A供体)处理的NRCM(新生大鼠心肌细胞)和接受TAC的Acad8cKO小鼠心脏进行RNA测序分析,发现心肌病相关基因和心脏肥大标志物的上调;TRANSFAC和JASPAR PWM分析表明TEAD2是两个RNA测序数据集中上调基因的转录因子。作者进一步在Nppa和Nppb的启动子区域内鉴定了潜在的TEAD2结合基序,并发现isobutyrate处理和Acad8敲低均显著增强了Nppa和Nppb启动子区域中的TEAD2结合。此外TEAD2过表达促进Acad8敲低和isobutyrate治疗诱导的Nppa表达。基于这些发现,作者推测异丁酰辅酶A和异丁酰化修饰的增加可能会促进染色质开放,从而增强肥大基因启动子处的TEAD2结合。因此作者评估了染色质可及性的变化,发现isobutyrate处理和Acad8敲低均导致Nppa和Nppb启动子区域染色质可及性增加。此外isobutyrate处理和Acad8敲低都促进了H3K9ibu和H3K23ibu在肥大相关基因Nppa启动子区域的富集。这些结果表明,Acad8缺陷通过异丁酰辅酶A的积累来调节组蛋白异丁酰化、染色质可及性和目标基因启动子区域TEAD2富集,从而导致心肌细胞肥大。
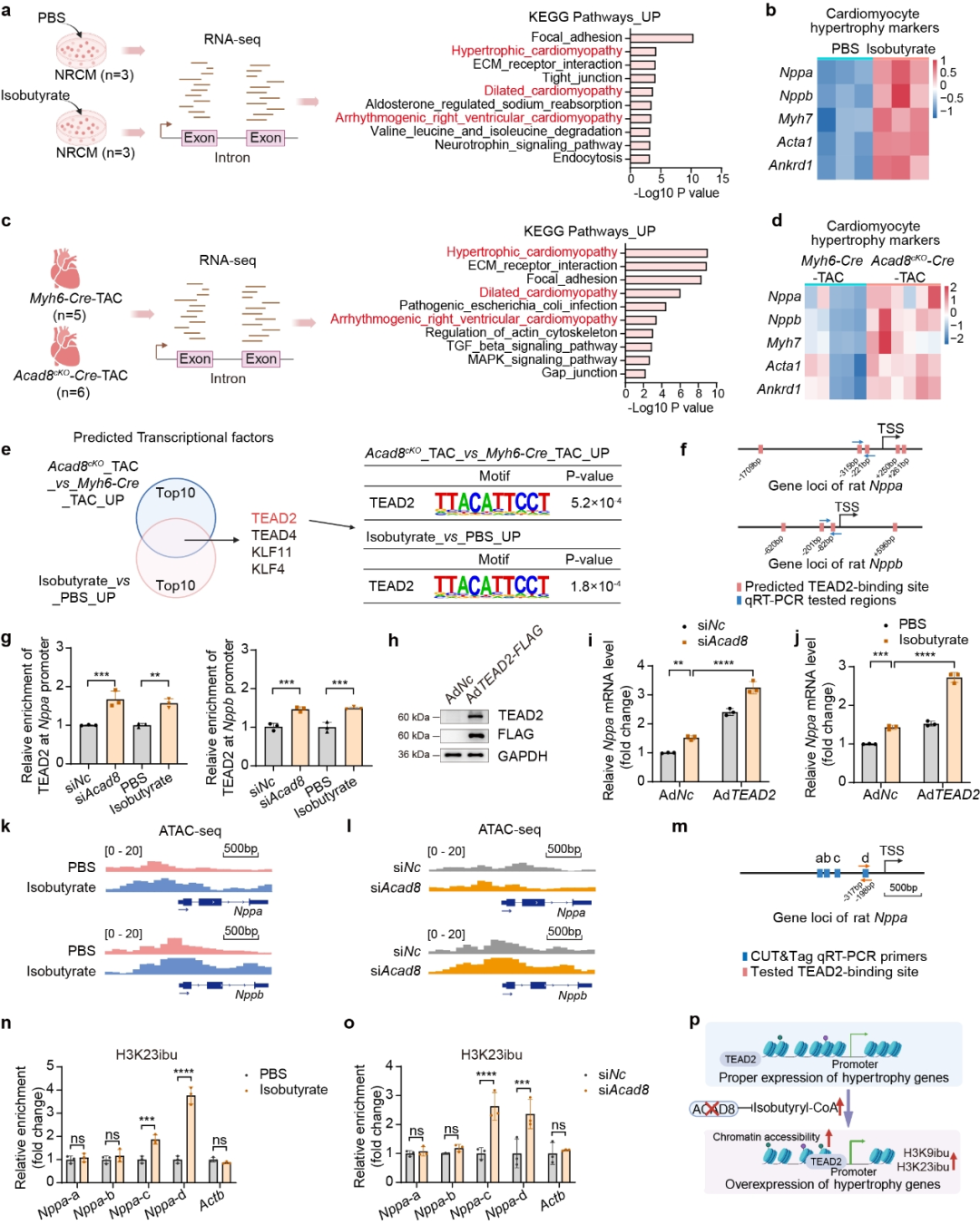
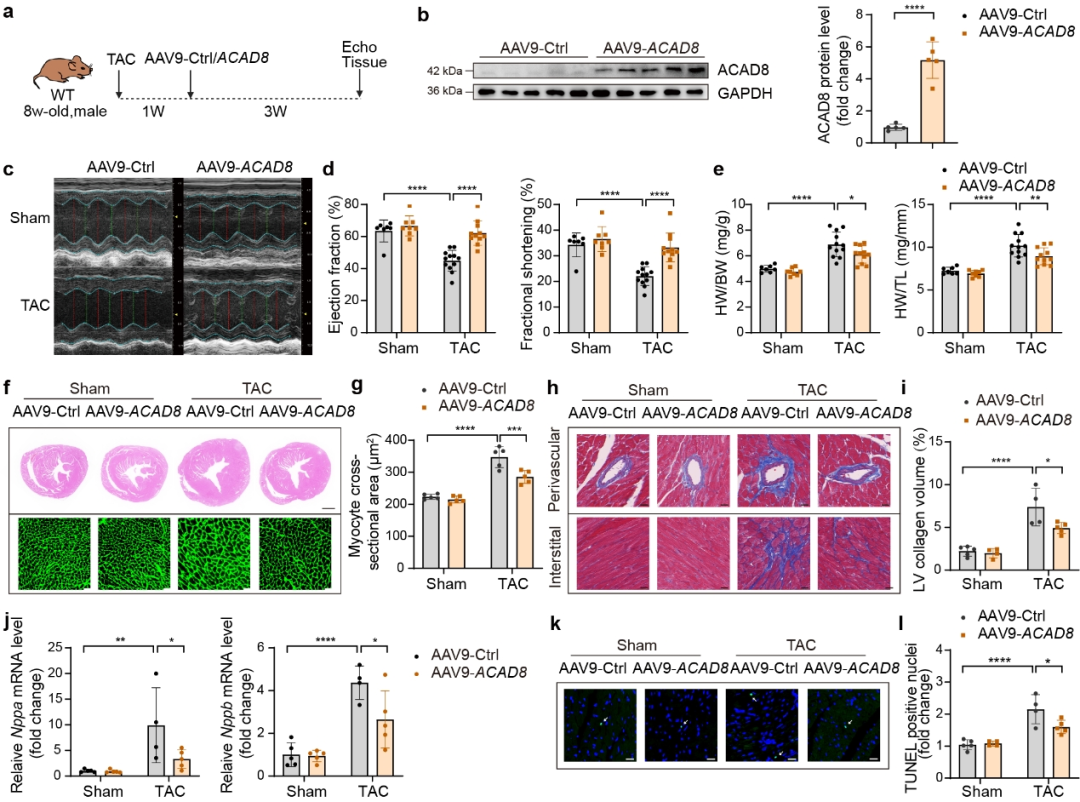
作者建立了TAC诱导的病理性心脏肥大的小鼠模型,并在TAC 7天后通过静脉注射AAV9-cTnT-Ctrl或AAV9-cTnT-ACAD8。结果显示心肌细胞特异性ACAD8过表达有效抑制TAC诱导的心脏功能障碍,减轻了心脏重量,减少了心脏肥大和纤维化以及肥大相关基因表达水平,减弱了TAC诱导的细胞死亡。这些发现表明心肌细胞特异性ACAD8过表达抑制TAC诱导的心脏肥大并恢复心脏功能。机制研究发现ACAD8过表达通过降低异丁酰辅酶A水平和组蛋白异丁酰化来改善病理性心脏肥大。
本研究强调了ACAD8在调节异丁酰辅酶A水平以维持心脏稳态中的作用,Acad8缺陷诱导的异丁酰辅酶A积累通过表观遗传重编程加剧了压力超负荷诱导的病理性心脏肥大。ACAD8-异丁酰辅酶A-异丁酰化轴可能是心脏肥大的新治疗靶点。

400-077-2566

service@wzbio.cn








