IF 9.0|山东中医药大学孔亮团队揭示PDK1在糖尿病神经病变中的神经保护机制
高血糖引起的糖代谢异常是糖尿病(DM)患者神经变性和认知功能障碍的一个致病因素。丙酮酸脱氢酶激酶(PDK)-乳酸轴被认为是代谢重编程与神经系统疾病发病过程之间的关键环节。然而,其在糖尿病性神经病变中的作用尚不清楚。2023年11月7日,山东中医药大学附属医院孔亮团队在Cell Death & Disease (IF 9.0) 发表题为“Pyruvate dehydrogenase kinase 1 protects against neuronal injury and memory loss in mouse models of diabetes”的研究论文。文章探讨了PDK1在高血糖诱导的异常代谢和神经元损伤中的作用和机制,乙酰辅酶A诱导的组蛋白乙酰化和HIF-1调控PDK1的表达,增强PDK1可能对糖尿病小鼠的认知恢复有保护作用。
研究方法与结果
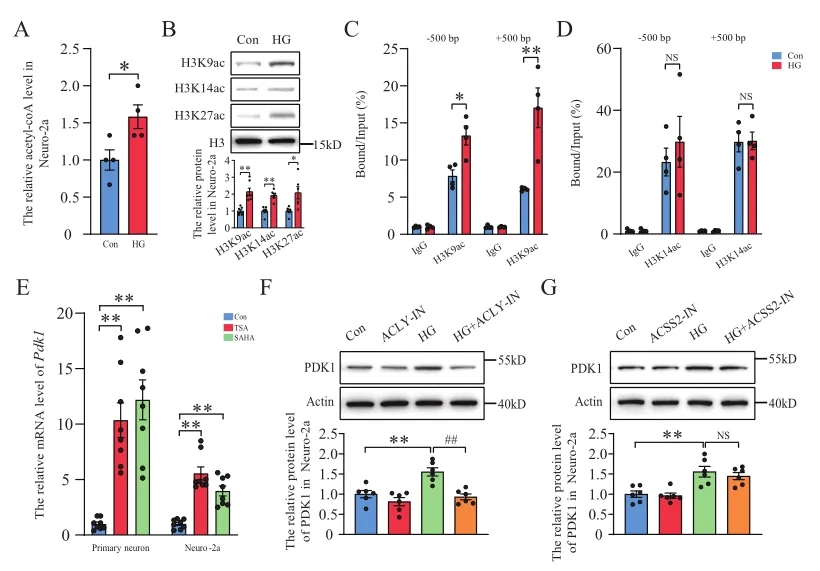
1、PDK1在糖尿病小鼠海马神经元中减少,在高糖处理的原代神经元中增加
以往研究表明,海马神经元损伤可能导致糖尿病小鼠记忆丧失。作者检测发现PDK1主要在神经元中表达,在糖尿病小鼠海马背侧(DH)的表达显著降低,但在高糖(HG)处理的原代神经元和Neuro-2a细胞中却显著增加。据报道,HG可以增加乙酰辅酶A诱导的系膜细胞组蛋白超乙酰化和基因表达水平,因此,作者研究了乙酰辅酶A诱导的组蛋白乙酰化对PDK1表达的影响。HG处理后,Pdk1基因启动子的H3K9乙酰化水平明显升高,使用组蛋白去乙酰化酶抑制剂TSA和SAHA处理均显著提高了原代神经元和Neuro-2a细胞中Pdk1的mRNA水平。以上数据表明HG刺激PDK1的表达增加可能依赖于乙酰辅酶A诱导的组蛋白乙酰化。进一步分析发现HIF-1参与HG诱导的Pdk1转录激活,并在糖尿病小鼠海马神经元中表达减少。
图1. HG刺激的神经元中,乙酰辅酶A诱导的组蛋白乙酰化介导PDK1的表达
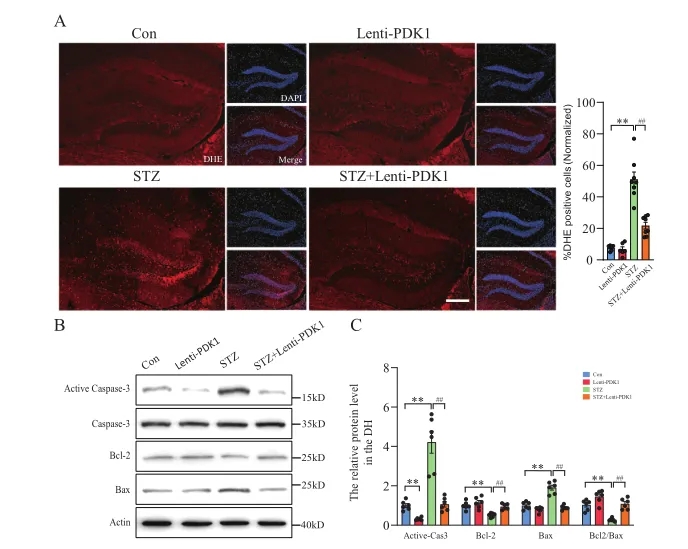
2、PDK1过表达可减轻海马ROS生成和神经元凋亡
为了探讨PDK1对HG诱导的氧化应激和神经元凋亡的影响,作者将Lenti-PDK1转染原代神经元和Neuro-2a细胞,PDK1的过表达明显抑制了HG诱导的氧化应激和神经元凋亡。接下来,作者研究了PDK1在糖尿病小鼠海马中的作用,小鼠体内注射Lenti-PDK1显著降低了STZ诱导的ROS释放,并逆转了STZ诱导的活性Caspase-3和Bax的增加以及Bcl-2的降低,表明PDK1对线粒体丙酮酸代谢的重编程可能减轻糖尿病高血糖诱导的线粒体ROS的产生,并进一步挽救海马神经元的凋亡。最后,作者通过对象识别记忆(ORM)和Morris水迷宫实验进一步证实海马PDK1具有预防高血糖诱导的神经元凋亡和改善糖尿病小鼠认知功能障碍的能力。
图2. PDK1减少STZ诱导的小鼠海马中ROS的产生和神经元凋亡
结论
本研究揭示了一种新的神经元自我保护机制,即乙酰辅酶A介导的组蛋白乙酰化直接上调PDK1,并通过抑制PDH活性来防止HG引发的线粒体代谢过载和氧化应激。然而,海马中HIF-1的减少损害了糖尿病小鼠PDK1的表达和神经元保护作用。文章揭示了PDK1在高血糖诱导的神经元凋亡和记忆丧失中的有效作用,提示PDK1-乳酸轴可作为治疗糖尿病神经病变的靶点。

 当前位置:首页 > 新闻中心 > 新闻资讯
当前位置:首页 > 新闻中心 > 新闻资讯